Das könnte Ihnen auch gefallen
- Aldehydes and Ketones - 1-MergedDokument94 SeitenAldehydes and Ketones - 1-MergedseNoch keine Bewertungen
- Scince Study MaterialDokument176 SeitenScince Study MaterialGAOCHU GAMERNoch keine Bewertungen
- Organic ChemistryDokument64 SeitenOrganic ChemistryJeetendra Yadav100% (1)
- DNA Structure and FunctionDokument15 SeitenDNA Structure and FunctionJomar Ison100% (2)
- Chamical Compound ListDokument7 SeitenChamical Compound ListMuni Reddy100% (1)
- Condensed Azines. Quinoline. Isoquinoline. Acridine. Diazines. Purine.Dokument151 SeitenCondensed Azines. Quinoline. Isoquinoline. Acridine. Diazines. Purine.omansuNoch keine Bewertungen
- Studies in Piperidone Chemistry. I. A Synthesis of 5-Homopiperazinones - J Org Chem, 1949, 14 (4), 530 - Jo01156a005Dokument7 SeitenStudies in Piperidone Chemistry. I. A Synthesis of 5-Homopiperazinones - J Org Chem, 1949, 14 (4), 530 - Jo01156a005muopioidreceptorNoch keine Bewertungen
- Test Bank For Biochemistry The Molecular Basis of Life 7th Edition James R Mckee Trudy MckeeDokument19 SeitenTest Bank For Biochemistry The Molecular Basis of Life 7th Edition James R Mckee Trudy Mckeeinflaterfloezuo86yNoch keine Bewertungen
- Recent Trends in Chemistry of Pyridine N-OxideDokument10 SeitenRecent Trends in Chemistry of Pyridine N-OxideNaresh KumarNoch keine Bewertungen
- Notes (CK), Koz-) : of 1,2-Dibrornoacenaphthylene-5,6-Sultone Acenaphthylene-5,6-SultoneDokument2 SeitenNotes (CK), Koz-) : of 1,2-Dibrornoacenaphthylene-5,6-Sultone Acenaphthylene-5,6-SultonetrungNoch keine Bewertungen
- Schulz1963 HCCOHDokument1 SeiteSchulz1963 HCCOHd20vipsplayNoch keine Bewertungen
- Bergel and Morrison 5-Hydroxyindole. 5-Hydroxyindole.: 5-Methoxyindole-2-Carboxylic 2-Nitro-5-HydroxytolueneDokument1 SeiteBergel and Morrison 5-Hydroxyindole. 5-Hydroxyindole.: 5-Methoxyindole-2-Carboxylic 2-Nitro-5-HydroxytolueneAnonymous FigYuONxuuNoch keine Bewertungen
- Piperidine Derivatives. XXI. 4-Piperidone, 4-Piperidinol and Certain of Their Derivatives - J Am Chem Soc, 1949, 71 (3), 901-906 - Ja01171a038Dokument6 SeitenPiperidine Derivatives. XXI. 4-Piperidone, 4-Piperidinol and Certain of Their Derivatives - J Am Chem Soc, 1949, 71 (3), 901-906 - Ja01171a038muopioidreceptorNoch keine Bewertungen
- PyridinesDokument14 SeitenPyridineskim haksongNoch keine Bewertungen
- NitrationDokument27 SeitenNitrationsubhashpithaniNoch keine Bewertungen
- 5231 The Synthesis of N Substituted Ureas I The N Alkylation of Ureas8647Dokument8 Seiten5231 The Synthesis of N Substituted Ureas I The N Alkylation of Ureas8647Chandra ShekharNoch keine Bewertungen
- ASSIGNMENT of Organic ChemistryDokument8 SeitenASSIGNMENT of Organic ChemistryWania AliNoch keine Bewertungen
- 1984 - WadsworthDkk - Enamines From Iodine Oxidation of Trialkylamines. 1. Electrophilic Capture by Cationic Heterocyclic RingsDokument6 Seiten1984 - WadsworthDkk - Enamines From Iodine Oxidation of Trialkylamines. 1. Electrophilic Capture by Cationic Heterocyclic RingsUAS TekimNoch keine Bewertungen
- And Of: Preparation Kinetics Decomposition of A Bicyclic Azo Compound. A Novel Reduction1Dokument6 SeitenAnd Of: Preparation Kinetics Decomposition of A Bicyclic Azo Compound. A Novel Reduction1itbwngNoch keine Bewertungen
- Amines Test Paper AnswerDokument12 SeitenAmines Test Paper AnswerprataprohanvnsNoch keine Bewertungen
- Clarkson1930 Primer SPFXDokument11 SeitenClarkson1930 Primer SPFXyulliarperezNoch keine Bewertungen
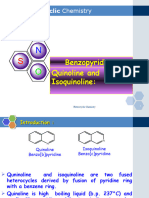
- Benzopyridines Quinoline and Isoquinoline:: Heterocyclic ChemistryDokument14 SeitenBenzopyridines Quinoline and Isoquinoline:: Heterocyclic ChemistryasfgegfNoch keine Bewertungen
- Reaction of Aminoquinones and Related Vinylogous Amides With Nitrous Acid. Synthesis and Chemistry of Cyclic Diazo KetonesDokument5 SeitenReaction of Aminoquinones and Related Vinylogous Amides With Nitrous Acid. Synthesis and Chemistry of Cyclic Diazo KetonesivanNoch keine Bewertungen
- A New Technique in Preparing 2,4-Dinitrophenylhydrazones.Dokument2 SeitenA New Technique in Preparing 2,4-Dinitrophenylhydrazones.H Vásquez GalindoNoch keine Bewertungen
- Arndt-Eistert Synthesis (Wolf Rearrangement) : O CH N N RC O OH R C O CL CH N 2 - Diazoketone R C O CH NNDokument54 SeitenArndt-Eistert Synthesis (Wolf Rearrangement) : O CH N N RC O OH R C O CL CH N 2 - Diazoketone R C O CH NNJosephine TorresNoch keine Bewertungen
- Chemistry NotesDokument16 SeitenChemistry NotesDipti GuptaNoch keine Bewertungen
- On 1 2 Benzisoxazole 3 Acetic AcidDokument2 SeitenOn 1 2 Benzisoxazole 3 Acetic AcidGI2015Noch keine Bewertungen
- Iron-Catalyzed C-N Bond Formations: Saisuree PrateeptongkumDokument70 SeitenIron-Catalyzed C-N Bond Formations: Saisuree Prateeptongkummarcel bokNoch keine Bewertungen
- Two Mark Questions With AnswersDokument5 SeitenTwo Mark Questions With AnswersHimani PavadiaNoch keine Bewertungen
- Otro Organocatalizador Coomo El de EVANS y TioureasDokument4 SeitenOtro Organocatalizador Coomo El de EVANS y TioureasFernando RSNoch keine Bewertungen
- Sales de Diazonio - ColorantesDokument12 SeitenSales de Diazonio - ColorantesnadiarhNoch keine Bewertungen
- Acid-Catalyzed Rearrangement of Some Aromatic Ketones'Dokument4 SeitenAcid-Catalyzed Rearrangement of Some Aromatic Ketones'Sandipan SahaNoch keine Bewertungen
- Unit - VDokument17 SeitenUnit - VSelva Mani100% (1)
- 2 Iodobenzoic AcidDokument4 Seiten2 Iodobenzoic AcidApril Ng0% (1)
- Name Reaction QuestionsDokument7 SeitenName Reaction Questionsakashkabiraj999Noch keine Bewertungen
- Recent Advances in The Synthesis of Pipe Rid Ones and Piperidines PM Weintraub JS Sabol JM Kane DR Borcherding Tetrahedron 59 2953 2989 2003Dokument37 SeitenRecent Advances in The Synthesis of Pipe Rid Ones and Piperidines PM Weintraub JS Sabol JM Kane DR Borcherding Tetrahedron 59 2953 2989 2003KybernetikumNoch keine Bewertungen
- Cyano Acetamide2Dokument2 SeitenCyano Acetamide2ashokNoch keine Bewertungen
- Capsaicin Synth - PHD - STRUCTURE AND BIOSYNTHESIS OF CAPSAICINDokument148 SeitenCapsaicin Synth - PHD - STRUCTURE AND BIOSYNTHESIS OF CAPSAICINyunusNoch keine Bewertungen
- Done By: Kaijie, Elias, Chenxi, Ashwini, Sahana, Kelly From 0901 and 0914 Compiled From 2007 and 2008 H2 Chemistry Prelim PapersDokument10 SeitenDone By: Kaijie, Elias, Chenxi, Ashwini, Sahana, Kelly From 0901 and 0914 Compiled From 2007 and 2008 H2 Chemistry Prelim Papersdiejunqs sNoch keine Bewertungen
- PhenolsDokument21 SeitenPhenolsdavid_tomy_10% (1)
- Hetero-Cyclic CompoundsDokument69 SeitenHetero-Cyclic CompoundsNaveed SajidNoch keine Bewertungen
- Professor Manihar THESISDokument183 SeitenProfessor Manihar THESISSatyabhama SanasamNoch keine Bewertungen
- Lec 1 RevisionDokument6 SeitenLec 1 RevisionadhamelthnNoch keine Bewertungen
- Catalytic Reduction With HydrazineDokument3 SeitenCatalytic Reduction With HydrazineOgnian DimitrovNoch keine Bewertungen
- 1982 - Catalytic Hydrocyanation of Dienes and TrienesDokument8 Seiten1982 - Catalytic Hydrocyanation of Dienes and TrienesJoão Augusto CruzNoch keine Bewertungen
- Water Effect On Air Oxidation ReactionsDokument5 SeitenWater Effect On Air Oxidation ReactionsSarat Na NiNoch keine Bewertungen
- Chiral Zinc Bipyridine Complex. Crystal Structure and Catalytic Activity in Asymmetric Allylation ReactionDokument4 SeitenChiral Zinc Bipyridine Complex. Crystal Structure and Catalytic Activity in Asymmetric Allylation ReactionMatefi Elena AlinaNoch keine Bewertungen
- Estudio de La CumarinaDokument5 SeitenEstudio de La CumarinaMarco Antonio MorenoNoch keine Bewertungen
- ExerciseDokument16 SeitenExercisecse.220840131017Noch keine Bewertungen
- Condensed Azines. Quinoline. Isoquinoline. Acridine. Diazines. Purine.Dokument151 SeitenCondensed Azines. Quinoline. Isoquinoline. Acridine. Diazines. Purine.Rajesh Kumar0% (2)
- The Kedzie Chemical Laboratory, Michigan Slate University, East Lansing, Mich. (U.S.A.)Dokument5 SeitenThe Kedzie Chemical Laboratory, Michigan Slate University, East Lansing, Mich. (U.S.A.)063Arifa Asya NasyidahNoch keine Bewertungen
- 2,4 DimethylazetidineDokument6 Seiten2,4 DimethylazetidineJana DittrichNoch keine Bewertungen
- Revised Organic Compounds Containing NitrogenDokument70 SeitenRevised Organic Compounds Containing NitrogenNabiAliNoch keine Bewertungen
- Organic and Biological Chemistry: Reaction of Iron Pentacarbonyl With Gem-DihalidesDokument4 SeitenOrganic and Biological Chemistry: Reaction of Iron Pentacarbonyl With Gem-DihalidesCabNoch keine Bewertungen
- Board Test - AldehydesDokument5 SeitenBoard Test - AldehydesMohammed IliasNoch keine Bewertungen
- NamereactionorganicDokument13 SeitenNamereactionorganicdeykrishna654100% (1)
- Tetrahedron: Tatiana B. Khlebnikova, Vasily N. Konev, Zinaida P. PaiDokument8 SeitenTetrahedron: Tatiana B. Khlebnikova, Vasily N. Konev, Zinaida P. Pai'Licenza AdagioNoch keine Bewertungen
- Aliphatics Home PackageDokument6 SeitenAliphatics Home PackageelishamahubiNoch keine Bewertungen
- Hydrocarbons Dps AssignmentDokument6 SeitenHydrocarbons Dps AssignmentharshillbhartiNoch keine Bewertungen
- Artt 1 PDFDokument4 SeitenArtt 1 PDFNicolás GrinbergNoch keine Bewertungen
- HydrocarbonsDokument30 SeitenHydrocarbonsSneha AunttyNoch keine Bewertungen
- Chapter 4Dokument21 SeitenChapter 4calvinabsNoch keine Bewertungen
- Silicon in Organic Synthesis: Butterworths Monographs in Chemistry and Chemical EngineeringVon EverandSilicon in Organic Synthesis: Butterworths Monographs in Chemistry and Chemical EngineeringNoch keine Bewertungen
- Transition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesVon EverandTransition Metal-Catalyzed Pyridine Synthesis: Transition Metal-Catalyzed Heterocycle Synthesis SeriesNoch keine Bewertungen
- Vitaminca CDokument5 SeitenVitaminca CJuanAmayaNoch keine Bewertungen
- Practica 4 - Curva CrecimientoDokument7 SeitenPractica 4 - Curva CrecimientoJuanAmayaNoch keine Bewertungen
- DryingDokument20 SeitenDryingJuanAmayaNoch keine Bewertungen
- Ezqui y DopaDokument7 SeitenEzqui y DopaJuanAmayaNoch keine Bewertungen
- A Acid Base ObjektifDokument3 SeitenA Acid Base ObjektifSaonah ZabaliNoch keine Bewertungen
- Organic Chemistry II / CHEM 252 Chapter 19 - Synthesis and: Reactions of β-Dicarbonyl CompoundsDokument40 SeitenOrganic Chemistry II / CHEM 252 Chapter 19 - Synthesis and: Reactions of β-Dicarbonyl CompoundsSilambarasan SivalingamNoch keine Bewertungen
- Coordination Chemistry of Chromium: PA Lay and A Levina, The University of Sydney, Sydney, AustraliaDokument50 SeitenCoordination Chemistry of Chromium: PA Lay and A Levina, The University of Sydney, Sydney, AustraliaAnjali ChauhanNoch keine Bewertungen
- Hard and Soft Acids and BasesDokument17 SeitenHard and Soft Acids and BasesSalmi Seprianti100% (2)
- Super Coils and Linking NumberDokument5 SeitenSuper Coils and Linking NumberShaher Bano MirzaNoch keine Bewertungen
- Metal ReductionDokument11 SeitenMetal Reductiondeepthimahanthi sabithaNoch keine Bewertungen
- BiuretDokument10 SeitenBiuretIndrie DwiraandaNoch keine Bewertungen
- 0620 - s13 - QP - 21 Igcse PaperDokument16 Seiten0620 - s13 - QP - 21 Igcse PaperGAURI GUPTANoch keine Bewertungen
- Lecture 7 Aromatic CompoundsDokument40 SeitenLecture 7 Aromatic CompoundsEricNoch keine Bewertungen
- Acids Bases and SaltsDokument4 SeitenAcids Bases and SaltsMandeep SinghNoch keine Bewertungen
- Hard and Soft Acid and BasesDokument15 SeitenHard and Soft Acid and BasesDr. Md. Ehtesham Ul Hoque100% (2)
- Pharmaceuticals - Library ContentDokument103 SeitenPharmaceuticals - Library ContentCH M RehanNoch keine Bewertungen
- CH 03Dokument308 SeitenCH 03Steven PaulNoch keine Bewertungen
- Physical Properties and Identification of Acid-Base Properties of Representative Organic Compounds Using Simple Solubility TestsDokument6 SeitenPhysical Properties and Identification of Acid-Base Properties of Representative Organic Compounds Using Simple Solubility TestsMatthew SA100% (1)
- Chemical Bonding: Why Bond Anyway?Dokument45 SeitenChemical Bonding: Why Bond Anyway?PutRi Charolin GintingNoch keine Bewertungen
- Grade 9 Chemistry Review ExerciseDokument6 SeitenGrade 9 Chemistry Review ExerciseJacqueline LaiNoch keine Bewertungen
- Basic Organic Nomenclature Packet Honors Chemistry: Name: - BlockDokument12 SeitenBasic Organic Nomenclature Packet Honors Chemistry: Name: - BlockJamaica Calamno SalvadorNoch keine Bewertungen
- Chem Academy: Chemical BondingDokument4 SeitenChem Academy: Chemical BondingEmraan EmmiNoch keine Bewertungen
- Synthesis and Characterization of New Complex Salts, of Some Transition and Non-Transition Metals With Isoquinolinium Derivative SaltsDokument7 SeitenSynthesis and Characterization of New Complex Salts, of Some Transition and Non-Transition Metals With Isoquinolinium Derivative SaltsIJRRRNoch keine Bewertungen
- Aromatic HydrocarbonsDokument15 SeitenAromatic HydrocarbonsasishNoch keine Bewertungen
- Basics of Ion ExchangeDokument4 SeitenBasics of Ion ExchangeRizka MaharanaNoch keine Bewertungen
- Disha Chemistry Revision (WWW - Crackjee.xyz)Dokument9 SeitenDisha Chemistry Revision (WWW - Crackjee.xyz)Tanmay Morey100% (1)
- Percent Composition & Hydrates & Empirical FormulasDokument2 SeitenPercent Composition & Hydrates & Empirical FormulasConnor BingamanNoch keine Bewertungen
- Acid-Base Concept WebDokument5 SeitenAcid-Base Concept WebSiow Kwong NieNoch keine Bewertungen
- Study Materials: Vedantu Innovations Pvt. Ltd. Score High With A Personal Teacher, Learn LIVE Online!Dokument12 SeitenStudy Materials: Vedantu Innovations Pvt. Ltd. Score High With A Personal Teacher, Learn LIVE Online!M. Amebari NongsiejNoch keine Bewertungen
- Coordination # and Ionic RadiiDokument9 SeitenCoordination # and Ionic RadiiAaila AkhterNoch keine Bewertungen