Das könnte Ihnen auch gefallen
- Neurophysiology of Depression and Bipolar Affective DisorderDokument17 SeitenNeurophysiology of Depression and Bipolar Affective DisorderShivan A.C.Noch keine Bewertungen
- Biological FactorsDokument3 SeitenBiological FactorsDEEPIKA PREMLAL 2170101Noch keine Bewertungen
- En A09v26n3 PDFDokument13 SeitenEn A09v26n3 PDFMilorad Miki AndjelkovicNoch keine Bewertungen
- Hypothalamic-Pituitary-Adrenal Axis and Bipolar DisorderDokument12 SeitenHypothalamic-Pituitary-Adrenal Axis and Bipolar DisorderCarolina MuñozNoch keine Bewertungen
- Serotonin and DepressionDokument5 SeitenSerotonin and Depressionsaad HussainNoch keine Bewertungen
- A Review: Hypothesis of Depression and Role of Antidepressant DrugsDokument4 SeitenA Review: Hypothesis of Depression and Role of Antidepressant DrugsSuraj SawadekarNoch keine Bewertungen
- Electro Convulsive TherapyDokument19 SeitenElectro Convulsive TherapyDeepshikha AhlawatNoch keine Bewertungen
- The Serotonin Hypothesis of Major DepressionDokument16 SeitenThe Serotonin Hypothesis of Major DepressionLorensia Fitra DwitaNoch keine Bewertungen
- Artigo Fisiopato DepressãoDokument21 SeitenArtigo Fisiopato DepressãoJ VichhyNoch keine Bewertungen
- Review Article Psychotropic-Induced Hyperprolactinemia: A Clinical ReviewDokument8 SeitenReview Article Psychotropic-Induced Hyperprolactinemia: A Clinical ReviewAndra SpatarNoch keine Bewertungen
- Lecture 2 (Psychotropic Drugs)Dokument17 SeitenLecture 2 (Psychotropic Drugs)ahmadslayman1Noch keine Bewertungen
- Is The Thyroid Still Important in Major Depression?: Editorial ÉditorialDokument2 SeitenIs The Thyroid Still Important in Major Depression?: Editorial ÉditorialAdina OlteanuNoch keine Bewertungen
- Both An Excess of Cortisol and DST Nonsuppression Have Been Reported For Many Years in Patients With Mood DisordersDokument12 SeitenBoth An Excess of Cortisol and DST Nonsuppression Have Been Reported For Many Years in Patients With Mood DisordersRubie Ann TillorNoch keine Bewertungen
- Movement DisordersDokument32 SeitenMovement DisordersMetta WitariNoch keine Bewertungen
- McCutcheon Et Al-2020-World PsychiatryDokument19 SeitenMcCutcheon Et Al-2020-World PsychiatryRob McCutcheonNoch keine Bewertungen
- Schizophrenia: Recent Advances and Future Hopes: by Ahmad Al-Dabbas Resident in PsychiatryDokument25 SeitenSchizophrenia: Recent Advances and Future Hopes: by Ahmad Al-Dabbas Resident in PsychiatryAhmad DabbasNoch keine Bewertungen
- HPA Axis in MDDDokument6 SeitenHPA Axis in MDDMarie SalomonováNoch keine Bewertungen
- Ezqui y DopaDokument7 SeitenEzqui y DopaJuanAmayaNoch keine Bewertungen
- Ukmss 32323Dokument19 SeitenUkmss 32323Paulo VianaNoch keine Bewertungen
- Documento TioDokument13 SeitenDocumento Tioxiomara velasquezNoch keine Bewertungen
- How Psychotherapy Changes The BrainDokument5 SeitenHow Psychotherapy Changes The BrainTomas SærmarkNoch keine Bewertungen
- Neuroendocrine Process in DepressionDokument3 SeitenNeuroendocrine Process in DepressionfarahNoch keine Bewertungen
- The Stress System in The Human Brain in Depression and NeurodegenerationDokument54 SeitenThe Stress System in The Human Brain in Depression and NeurodegenerationRomeo IsaíasNoch keine Bewertungen
- The Pathophysiology and Genetics of OCDDokument12 SeitenThe Pathophysiology and Genetics of OCDCrescent FangNoch keine Bewertungen
- PV Interneuron Alterations in Stress RelDokument98 SeitenPV Interneuron Alterations in Stress RelSea SaltNoch keine Bewertungen
- Vidal 2011Dokument13 SeitenVidal 2011xoxomeNoch keine Bewertungen
- TAHONLAMA Co-Administration of Sertraline and HaloperidolDokument6 SeitenTAHONLAMA Co-Administration of Sertraline and Haloperidolmijon46Noch keine Bewertungen
- Chapter 5 - Depression - 2018 - Integrative MedicineDokument13 SeitenChapter 5 - Depression - 2018 - Integrative MedicineandreNoch keine Bewertungen
- 2018 - ARTIGO - Recognizing Depression From The Microbita Gut Axis - 8 Paginas (Tudo)Dokument16 Seiten2018 - ARTIGO - Recognizing Depression From The Microbita Gut Axis - 8 Paginas (Tudo)Liliana de SousaNoch keine Bewertungen
- Aging of The Endocrine System: Ieva B. AkbarDokument41 SeitenAging of The Endocrine System: Ieva B. AkbarvansrodNoch keine Bewertungen
- Efficacy and Safety of The 5-Hydroxytryptophan On Depression and Apathy inDokument18 SeitenEfficacy and Safety of The 5-Hydroxytryptophan On Depression and Apathy inIrma DanielleNoch keine Bewertungen
- Michelle Gillis - Phenylethylamine: More Than Just A Pea-Sized NeurochemicalDokument3 SeitenMichelle Gillis - Phenylethylamine: More Than Just A Pea-Sized NeurochemicalNeerFamNoch keine Bewertungen
- 272ano Farmacologia Principles of PharmacologyDokument27 Seiten272ano Farmacologia Principles of PharmacologyJoana CarvalhoNoch keine Bewertungen
- By Timothy R. Test, SR., Ph.D. National College Everest College of PhoenixDokument32 SeitenBy Timothy R. Test, SR., Ph.D. National College Everest College of PhoenixTimothy Robert TestNoch keine Bewertungen
- Mechanisms Involved in Placebo and Nocebo Responses and Implications For Drug TrialsDokument5 SeitenMechanisms Involved in Placebo and Nocebo Responses and Implications For Drug TrialsAlfredo GarciaNoch keine Bewertungen
- Inhibidores Recep Serotonina 2Dokument14 SeitenInhibidores Recep Serotonina 2FarmaFMNoch keine Bewertungen
- Serotonin & MigraineDokument10 SeitenSerotonin & Migrainepetri_jvNoch keine Bewertungen
- tmpE99B TMPDokument16 SeitentmpE99B TMPFrontiersNoch keine Bewertungen
- Etiology of OCD: Chairperson: DR Anupama Presenter: DR ChandiniDokument35 SeitenEtiology of OCD: Chairperson: DR Anupama Presenter: DR Chandinidrkadiyala2Noch keine Bewertungen
- Biology of DepressionDokument5 SeitenBiology of DepressionScience RespondsNoch keine Bewertungen
- Psilocybin Induces Schizophrenia-Like Psychosis in Humans Via A Serotonin-2 Agonist ActionDokument8 SeitenPsilocybin Induces Schizophrenia-Like Psychosis in Humans Via A Serotonin-2 Agonist ActionluthaisNoch keine Bewertungen
- Role of Thyroid Hormone Therapy in Depressive Disorders Bauer2021Dokument7 SeitenRole of Thyroid Hormone Therapy in Depressive Disorders Bauer2021fullscallopNoch keine Bewertungen
- New Insights Into Sympathetic Regulation of Glucose and Fat MetabolismDokument17 SeitenNew Insights Into Sympathetic Regulation of Glucose and Fat MetabolismAli HaidarNoch keine Bewertungen
- Molecular NeurobiologyDokument17 SeitenMolecular NeurobiologypairednursingNoch keine Bewertungen
- SULPYCO Method: A New Quantum and Integrative Approach to DepressionVon EverandSULPYCO Method: A New Quantum and Integrative Approach to DepressionNoch keine Bewertungen
- Therapeutics II - 1Dokument9 SeitenTherapeutics II - 1shukranamer2Noch keine Bewertungen
- Serotonina y Depresioì N 1 PDFDokument8 SeitenSerotonina y Depresioì N 1 PDFJenniferNoch keine Bewertungen
- Franz X. Vollenweider and Mark A. Geyer - A Systems Model of Altered Consciousness: Integrating Natural and Drug-Induced PsychosesDokument13 SeitenFranz X. Vollenweider and Mark A. Geyer - A Systems Model of Altered Consciousness: Integrating Natural and Drug-Induced PsychosesCortate15gNoch keine Bewertungen
- Reversal of Cocaine Addiction by Environmental EnrichmentDokument2 SeitenReversal of Cocaine Addiction by Environmental EnrichmentdsafasdfsdafaNoch keine Bewertungen
- Glutamate Neurotransmission in Psychotic Disorders and Substance AbuseDokument8 SeitenGlutamate Neurotransmission in Psychotic Disorders and Substance AbuseLeanne MontgomeryNoch keine Bewertungen
- Neurobiologi Depresi 2000 WFDokument6 SeitenNeurobiologi Depresi 2000 WFKim AntelNoch keine Bewertungen
- The Underlying Neurobiology of Bipolar Disorder: H K. M, J A. Q, J L. P, J S, B P. L, J S. V, C A. ZDokument11 SeitenThe Underlying Neurobiology of Bipolar Disorder: H K. M, J A. Q, J L. P, J S, B P. L, J S. V, C A. ZRhodis PeñaNoch keine Bewertungen
- Pharmacology of AutacoidsDokument13 SeitenPharmacology of AutacoidsInocenteNoch keine Bewertungen
- Pilhatsch 2011Dokument7 SeitenPilhatsch 2011omar rodriguezNoch keine Bewertungen
- DocumentDokument5 SeitenDocumentTop MusicNoch keine Bewertungen
- Chidomedscape - Delirium - Practice Essentials, Background, PathophysiologyDokument2 SeitenChidomedscape - Delirium - Practice Essentials, Background, PathophysiologyMikeVDCNoch keine Bewertungen
- Interface Between Hypothalamic-Pituitary-Adrenal Axis Andbrain-Derived Neurotrophic Factor in DepressionDokument13 SeitenInterface Between Hypothalamic-Pituitary-Adrenal Axis Andbrain-Derived Neurotrophic Factor in DepressioncarlosNoch keine Bewertungen
- Cancer Cachexia-Pathophysiology and ManagementDokument21 SeitenCancer Cachexia-Pathophysiology and ManagementDianne Faye ManabatNoch keine Bewertungen
- Siegfried 1993Dokument2 SeitenSiegfried 1993Haseeba KhanNoch keine Bewertungen
- Mood Disorders Lecture 2Dokument10 SeitenMood Disorders Lecture 2DEEPIKA PREMLAL 2170101Noch keine Bewertungen
- Ia201604s2-A18 87Dokument2 SeitenIa201604s2-A18 87Sri DewiNoch keine Bewertungen
- JurnalDokument3 SeitenJurnalSri DewiNoch keine Bewertungen
- JurnalDokument21 SeitenJurnalSri DewiNoch keine Bewertungen
- Reaksi Subtitusi Asil Nukleofilik Pada Turunan Asam KarboksilatDokument24 SeitenReaksi Subtitusi Asil Nukleofilik Pada Turunan Asam KarboksilatSri DewiNoch keine Bewertungen
- Leoline Installation and MaintenanceDokument8 SeitenLeoline Installation and MaintenanceFloorkitNoch keine Bewertungen
- Comparative Study of DEA and MDEADokument4 SeitenComparative Study of DEA and MDEAsaleh4060Noch keine Bewertungen
- Put The Verbs in Brackets Into The - Ing Form or The InfinitiveDokument10 SeitenPut The Verbs in Brackets Into The - Ing Form or The InfinitiveThao DaoNoch keine Bewertungen
- Lecture 38Dokument10 SeitenLecture 38Deepak GuptaNoch keine Bewertungen
- ST. LUKE'S MEDICAL CENTER EMPLOYEE'S FOUNDATION AFW v. NLRCDokument3 SeitenST. LUKE'S MEDICAL CENTER EMPLOYEE'S FOUNDATION AFW v. NLRCjodelle11Noch keine Bewertungen
- Summary of Germ LayersDokument2 SeitenSummary of Germ Layersaichiii.bearNoch keine Bewertungen
- MPCA Response 5.29.20Dokument2 SeitenMPCA Response 5.29.20Duluth News TribuneNoch keine Bewertungen
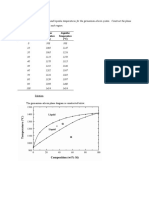
- Composition Solidus Temperature Liquidus Temperature: (WT% Si) (°C) (°C)Dokument7 SeitenComposition Solidus Temperature Liquidus Temperature: (WT% Si) (°C) (°C)Muhammad Ibkar YusranNoch keine Bewertungen
- ENT Head and Neck ExamDokument21 SeitenENT Head and Neck ExamvickyNoch keine Bewertungen
- CAST Optical Smoke Detector: Part NoDokument3 SeitenCAST Optical Smoke Detector: Part NoAnis TantoushNoch keine Bewertungen
- MSW TACSOP v.3.3Dokument59 SeitenMSW TACSOP v.3.3Mira BellaNoch keine Bewertungen
- Formula Sheet Pre-MidDokument4 SeitenFormula Sheet Pre-MidUzair KhanNoch keine Bewertungen
- BIS Ventilation Brochure enDokument16 SeitenBIS Ventilation Brochure enBruno SantosNoch keine Bewertungen
- Final Workshop Report On Value Addition and AgroprocessingDokument31 SeitenFinal Workshop Report On Value Addition and AgroprocessingBett K. BernardNoch keine Bewertungen
- VEIKK A15PRO Instruction Manual 0714Dokument20 SeitenVEIKK A15PRO Instruction Manual 0714Corny777 UwUNoch keine Bewertungen
- LabStan - July 16-17, 23-24, 30-31, Aug 6-7Dokument25 SeitenLabStan - July 16-17, 23-24, 30-31, Aug 6-7CandypopNoch keine Bewertungen
- L-6th Sem (Eng Notes) Law Relating To Women and ChildDokument52 SeitenL-6th Sem (Eng Notes) Law Relating To Women and ChildCuriae corporate consultantsNoch keine Bewertungen
- Parenting Styles and Social Interaction of Senior Secondary School Students in Imo State, NigeriaDokument10 SeitenParenting Styles and Social Interaction of Senior Secondary School Students in Imo State, NigeriaInternational Educational Applied Scientific Research Journal (IEASRJ)Noch keine Bewertungen
- Using The Time Domain Reflectometer To Check For and Locate A FaultDokument5 SeitenUsing The Time Domain Reflectometer To Check For and Locate A FaultSikandar MasoodNoch keine Bewertungen
- Mechanism of Enzyme ActionDokument19 SeitenMechanism of Enzyme ActionRubi AnnNoch keine Bewertungen
- Winchester Model 9422 Lever Action Rifle Owner's Manual: LicenseeDokument0 SeitenWinchester Model 9422 Lever Action Rifle Owner's Manual: Licenseecarlosfanjul1Noch keine Bewertungen
- Cell Division-Mitosis Notes: 2 New CellsDokument21 SeitenCell Division-Mitosis Notes: 2 New CellsCristina MariaNoch keine Bewertungen
- All Electricity Worksheets For Midterm 22Dokument22 SeitenAll Electricity Worksheets For Midterm 22Maryam AlkaabiNoch keine Bewertungen
- Fourier Ptychography Stivi ElbiDokument20 SeitenFourier Ptychography Stivi ElbistiviNoch keine Bewertungen
- Michigan Clinic 2008 NotesDokument10 SeitenMichigan Clinic 2008 NotesCoach Brown100% (3)
- Chapter 4 - Medical Aspects of Food SafetyDokument17 SeitenChapter 4 - Medical Aspects of Food SafetyasushkNoch keine Bewertungen
- Predictive Ability of Social Cognitive Theory in Exercise Research: An Integrated Literature ReviewDokument13 SeitenPredictive Ability of Social Cognitive Theory in Exercise Research: An Integrated Literature ReviewJovenlou BihagNoch keine Bewertungen
- Supplementary Spec To API Specification 17D Subsea Wellhead and Tree Equipment With Justifications S 561Jv2022 11Dokument81 SeitenSupplementary Spec To API Specification 17D Subsea Wellhead and Tree Equipment With Justifications S 561Jv2022 11maximusala83Noch keine Bewertungen
- Increase Credit Limit PDFDokument1 SeiteIncrease Credit Limit PDFemc2_mcvNoch keine Bewertungen
- Kmartinez Draft Research PaperDokument14 SeitenKmartinez Draft Research Paperapi-273007806Noch keine Bewertungen