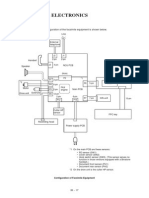
Das könnte Ihnen auch gefallen
- Patton Line Size - 2014Dokument6 SeitenPatton Line Size - 2014Rocky James AsildoNoch keine Bewertungen
- Introduction To Econometrics - Stock & Watson - CH 13 SlidesDokument38 SeitenIntroduction To Econometrics - Stock & Watson - CH 13 SlidesAntonio AlvinoNoch keine Bewertungen
- 5 3-6 EALD Progression by Mode SpeakingDokument1 Seite5 3-6 EALD Progression by Mode SpeakingS TANCREDNoch keine Bewertungen
- Demag Cranes CatalogDokument20 SeitenDemag Cranes Catalogmadhukarreddy2811100% (2)
- ht56r6x 6xxv110Dokument108 Seitenht56r6x 6xxv110tanhaoclcNoch keine Bewertungen
- Generadores de Vacío: Serie MICRO-SchmalzDokument5 SeitenGeneradores de Vacío: Serie MICRO-SchmalzHoracio CalderonNoch keine Bewertungen
- RailRoadCar Rearrangement IteratorDokument8 SeitenRailRoadCar Rearrangement IteratorRebel Sangamesh HNoch keine Bewertungen
- Grelhas de Retorno: AT e VAT: Tabelas de SeleçãoDokument2 SeitenGrelhas de Retorno: AT e VAT: Tabelas de SeleçãoRaphael LopesNoch keine Bewertungen
- VLAN Trunking Protocol (VTP) : Nexus Technology Labs Classical Ethernet SwitchingDokument6 SeitenVLAN Trunking Protocol (VTP) : Nexus Technology Labs Classical Ethernet SwitchingsatourismNoch keine Bewertungen
- Ads 1232 ReferenceDokument31 SeitenAds 1232 ReferenceMochammad SofyanNoch keine Bewertungen
- Question File PDFDokument23 SeitenQuestion File PDFJyothi Swaroop AllaNoch keine Bewertungen
- Lec 5Dokument22 SeitenLec 5Ahmed FarhoudNoch keine Bewertungen
- DiffusionReaction EXCEL FPDokument9 SeitenDiffusionReaction EXCEL FPLong TranNoch keine Bewertungen
- Buloneria - CatalogoDokument14 SeitenBuloneria - CatalogoJacinto LopezNoch keine Bewertungen
- Buloneria - CatalogoDokument14 SeitenBuloneria - CatalogoJacinto LopezNoch keine Bewertungen
- Buloneria - CatalogoDokument14 SeitenBuloneria - CatalogoJacinto LopezNoch keine Bewertungen
- Buloneria - CatalogoDokument14 SeitenBuloneria - CatalogoJacinto LopezNoch keine Bewertungen
- U2 Medley: Doyle DykesDokument15 SeitenU2 Medley: Doyle DykessuccamehuNoch keine Bewertungen
- Control Housing, Removal and Installation PDFDokument1 SeiteControl Housing, Removal and Installation PDFjustyn robergeNoch keine Bewertungen
- Year 5 Autumn Block 2 Step 2 HW EXT Subtract More Than 4 DigitsDokument5 SeitenYear 5 Autumn Block 2 Step 2 HW EXT Subtract More Than 4 DigitsNehal IsmailNoch keine Bewertungen
- Using 74LS138 IC to implement 5-to-32 decoder and 7-segment display problemsDokument5 SeitenUsing 74LS138 IC to implement 5-to-32 decoder and 7-segment display problemshqthang19952602Noch keine Bewertungen
- BKT Power Distribution SystemsDokument23 SeitenBKT Power Distribution SystemsGicuNoch keine Bewertungen
- Aim Neet Part Test 18Dokument22 SeitenAim Neet Part Test 18Hemant KumarNoch keine Bewertungen
- Bosch Rexroth - Catalog de Produse de BazăDokument38 SeitenBosch Rexroth - Catalog de Produse de Bazăjo_rz_57Noch keine Bewertungen
- Nintendo Wii UDokument2 SeitenNintendo Wii UNathalie PresillasNoch keine Bewertungen
- Chimp Spanner - Harvey WallbangerDokument8 SeitenChimp Spanner - Harvey WallbangerKenneth CastilloNoch keine Bewertungen
- Salida Terminal 20 Jun 2023Dokument67 SeitenSalida Terminal 20 Jun 2023Milder Jhaic LombardoNoch keine Bewertungen
- Aim Neet Part Test 08Dokument22 SeitenAim Neet Part Test 08piyushsharmaproductivityNoch keine Bewertungen
- Neet Test Series 2022 Test Code: NT - 0 4: Biology Physics ChemistryDokument23 SeitenNeet Test Series 2022 Test Code: NT - 0 4: Biology Physics Chemistry6 months AgoNoch keine Bewertungen
- Neet Test Series 2022 Test Code: NT - 01: Biology Physics ChemistryDokument17 SeitenNeet Test Series 2022 Test Code: NT - 01: Biology Physics Chemistry6 months AgoNoch keine Bewertungen
- Neet Test Series 2022 Test Code: NT - 02: Biology Physics ChemistryDokument16 SeitenNeet Test Series 2022 Test Code: NT - 02: Biology Physics Chemistry6 months AgoNoch keine Bewertungen
- Neet Test Series 2022 Test Code: NT - 12: Biology Physics ChemistryDokument23 SeitenNeet Test Series 2022 Test Code: NT - 12: Biology Physics Chemistry꧁༒ŞɨĐĐHÄRŦ卄༒꧂ ᴊʜᴀ࿐Noch keine Bewertungen
- Neet Test Series 2022 Test Code: NT - 0 6: Biology Physics ChemistryDokument22 SeitenNeet Test Series 2022 Test Code: NT - 0 6: Biology Physics ChemistryEmily MaxwellNoch keine Bewertungen
- Aim Neet Part Test 17Dokument25 SeitenAim Neet Part Test 17Mohammad saifNoch keine Bewertungen
- NEET Test Series 2022Dokument22 SeitenNEET Test Series 20226 months AgoNoch keine Bewertungen
- Instruction Latencies and Throughput For AMD and Intel x86 ProcessorsDokument7 SeitenInstruction Latencies and Throughput For AMD and Intel x86 Processorsgeorge_pitichNoch keine Bewertungen
- MatrizDokument1 SeiteMatrizFélix Alfredo Carpio CardonaNoch keine Bewertungen
- Performance of The Taylor Series Method For Odes/Daes: Roberto BarrioDokument21 SeitenPerformance of The Taylor Series Method For Odes/Daes: Roberto BarrioUzoma Nnaemeka TeflondonNoch keine Bewertungen
- LetrasDokument2 SeitenLetrasteacherrociocomeinschoolNoch keine Bewertungen
- Admission Form for Engineering Entrance ExamDokument2 SeitenAdmission Form for Engineering Entrance ExamDevyani GangwarNoch keine Bewertungen
- Target PBDDokument10 SeitenTarget PBDCHANDRAKUMARI A/P NADARAJAH MoeNoch keine Bewertungen
- Neet Test Series 2022 Test Code: NT - 14: Biology Physics ChemistryDokument20 SeitenNeet Test Series 2022 Test Code: NT - 14: Biology Physics ChemistrySeerat JanNoch keine Bewertungen
- Brother 190 SM 04Dokument19 SeitenBrother 190 SM 04Sunny SinghNoch keine Bewertungen
- Glastic Standoffs InsulatorsDokument2 SeitenGlastic Standoffs Insulatorsdanielliram993Noch keine Bewertungen
- CienDokument4 SeitenCienRafael CardosoNoch keine Bewertungen
- Neet Test Series 2022 Test Code: NT - 0 3: Biology Physics ChemistryDokument23 SeitenNeet Test Series 2022 Test Code: NT - 0 3: Biology Physics ChemistrySk jatuuNoch keine Bewertungen
- Density Based Traffic Light Control SystemDokument34 SeitenDensity Based Traffic Light Control SystemPrabhakarSinghNoch keine Bewertungen
- Drying Oil ProductionDokument21 SeitenDrying Oil ProductionAbdulla S. MahdiNoch keine Bewertungen
- Guidelines For Report Writing G78/G84/R75 Project Work ReportDokument14 SeitenGuidelines For Report Writing G78/G84/R75 Project Work ReportSyed Ahmed KabeerNoch keine Bewertungen
- T2 Selection and Installation GuideDokument1 SeiteT2 Selection and Installation GuideSamer ChahroukNoch keine Bewertungen
- Vignan Project HRDokument41 SeitenVignan Project HRMoin DugguNoch keine Bewertungen
- Place Value Chart Odd or Even Comparing NumbersDokument12 SeitenPlace Value Chart Odd or Even Comparing NumbersJenny Himor BalitaNoch keine Bewertungen
- 3. Programming Assignment From Chapter 1 to 6Dokument3 Seiten3. Programming Assignment From Chapter 1 to 6Mayuri PopatNoch keine Bewertungen
- Country Project Directions 2014Dokument10 SeitenCountry Project Directions 2014api-247673978Noch keine Bewertungen
- WebernOp27 II ProjDokument3 SeitenWebernOp27 II Projyeh30426Noch keine Bewertungen
- Vazadores (Punções) : E1. Cortecentral, Altura23,8 MM U N I D: 2 5 P Eç AsDokument4 SeitenVazadores (Punções) : E1. Cortecentral, Altura23,8 MM U N I D: 2 5 P Eç AsLeonardoNoch keine Bewertungen
- 1/16 Scale Models - Special Sets: PZ - Kpfw. VI, Tiger I, Ausf.E (SD - Kfz. 181) - Early VersionDokument5 Seiten1/16 Scale Models - Special Sets: PZ - Kpfw. VI, Tiger I, Ausf.E (SD - Kfz. 181) - Early VersionsharkmouthNoch keine Bewertungen
- BT_ExcelDokument27 SeitenBT_Excelthydung20042811Noch keine Bewertungen
- Auto Electronics Projects: An Introduction to Your Car Electrics with Useful and Proven Self-Buld ProjectsVon EverandAuto Electronics Projects: An Introduction to Your Car Electrics with Useful and Proven Self-Buld ProjectsBewertung: 5 von 5 Sternen5/5 (1)
- Sunday Worship ServiceDokument19 SeitenSunday Worship ServiceKathleen OlmillaNoch keine Bewertungen
- Lord of The Flies Unit PlanDokument28 SeitenLord of The Flies Unit Planapi-312430378100% (2)
- Exact Differential EquationsDokument7 SeitenExact Differential EquationsnimbuumasalaNoch keine Bewertungen
- Problem Set 2Dokument4 SeitenProblem Set 2Thomas LimNoch keine Bewertungen
- Module 3 Eng 107 Teaching and Assessment of LITDokument2 SeitenModule 3 Eng 107 Teaching and Assessment of LITJohlian TajanlangitNoch keine Bewertungen
- Translationand CensorshipDokument30 SeitenTranslationand CensorshipmozaroliveiraNoch keine Bewertungen
- Musical DevicesDokument26 SeitenMusical DevicesDave Supat TolentinoNoch keine Bewertungen
- TM Functional Safety 07 2011 enDokument229 SeitenTM Functional Safety 07 2011 enWaldemarNoch keine Bewertungen
- Japanese Literature As World Literature - Visceral Engagement PDFDokument253 SeitenJapanese Literature As World Literature - Visceral Engagement PDFMonica TamașNoch keine Bewertungen
- Spectrum Archive VMDokument83 SeitenSpectrum Archive VMMahendraNoch keine Bewertungen
- 3B Laugeson - The Science of Making FriendsDokument47 Seiten3B Laugeson - The Science of Making Friendslucia estrellaNoch keine Bewertungen
- Completing Sentences Using Given WordsDokument4 SeitenCompleting Sentences Using Given WordsTante DeviNoch keine Bewertungen
- Religion and Politics in EthiopiaDokument26 SeitenReligion and Politics in EthiopiaAbduljelil Sheh Ali KassaNoch keine Bewertungen
- TrianglesDokument139 SeitenTrianglessujaritha sureshNoch keine Bewertungen
- Meeting 2 PRONOUNSDokument9 SeitenMeeting 2 PRONOUNShappy heriesnNoch keine Bewertungen
- Advantages Disadvantages of DbmsDokument3 SeitenAdvantages Disadvantages of DbmsdunnkamwiNoch keine Bewertungen
- The Parable of Rainbow Colors - Actual 17Dokument6 SeitenThe Parable of Rainbow Colors - Actual 17Mark Lagorra Perez25% (4)
- Tu Ingles Sesion 22 TranscripcionDokument8 SeitenTu Ingles Sesion 22 TranscripcionEliaz MendezNoch keine Bewertungen
- Evaluation Test 6-Unit IDokument2 SeitenEvaluation Test 6-Unit IРотари ДианаNoch keine Bewertungen
- MTH401 #Zero Lecture: Discrete MathematicsDokument25 SeitenMTH401 #Zero Lecture: Discrete MathematicsPubg CloudNoch keine Bewertungen
- MavenDokument28 SeitenMavenniraj chavhanNoch keine Bewertungen
- Sharah Aqeeda-E-Tahawiyyah - Shaykh Muhammad Bin Yahya NinowyDokument205 SeitenSharah Aqeeda-E-Tahawiyyah - Shaykh Muhammad Bin Yahya NinowyAtif Hussain Khan100% (2)
- Passive VoiceDokument2 SeitenPassive VoiceArni Dwi astutiNoch keine Bewertungen
- Managing 15000 Network Devices With AnsibleDokument23 SeitenManaging 15000 Network Devices With AnsibleAshwini Kumar100% (1)
- Manuel S. Enverga University Foundation Lucena City Granted Autonomous Status CHED CEB Res. 076-2009 College of Arts and SciencesDokument3 SeitenManuel S. Enverga University Foundation Lucena City Granted Autonomous Status CHED CEB Res. 076-2009 College of Arts and SciencesLaika MerleNoch keine Bewertungen
- Java SerializationDokument3 SeitenJava SerializationVishnu Teerthacharya IbharampurNoch keine Bewertungen
- Build A Bootable BCD From Scratch With BcdeditDokument2 SeitenBuild A Bootable BCD From Scratch With BcdeditNandrianina RandrianarisonNoch keine Bewertungen
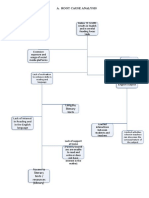
- Project REAP-Root Cause AnalysisDokument4 SeitenProject REAP-Root Cause AnalysisAIRA NINA COSICONoch keine Bewertungen
- Group B Assignment: File Transfer Using UDP SocketsDokument5 SeitenGroup B Assignment: File Transfer Using UDP Socketsvaibhav patilNoch keine Bewertungen