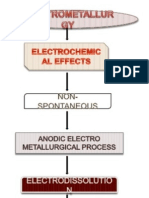
Das könnte Ihnen auch gefallen
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (122)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (590)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (842)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5807)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (401)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (897)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1091)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Electro-Polishing: Prepared byDokument44 SeitenElectro-Polishing: Prepared byMahesh Kumar100% (3)
- Electrochemistry Lecture NotesDokument28 SeitenElectrochemistry Lecture NotesRanjith Thamizhan100% (4)
- J Seppur 2018 12 040Dokument48 SeitenJ Seppur 2018 12 040SHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- J Scitotenv 2020 142108Dokument26 SeitenJ Scitotenv 2020 142108SHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Decoding The Pathways of Arsenic Biotransformation in BacteriaDokument24 SeitenDecoding The Pathways of Arsenic Biotransformation in BacteriaSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- J Apcatb 2019 02 041 PDFDokument24 SeitenJ Apcatb 2019 02 041 PDFSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Teoria de Oxidacion Avanzada Muy BuenaDokument9 SeitenTeoria de Oxidacion Avanzada Muy BuenaSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Arsenic Removal From Tap Water by Electrocoagulation: Investigation of Process Parameters, Kinetic Analysis, and Operating CostDokument11 SeitenArsenic Removal From Tap Water by Electrocoagulation: Investigation of Process Parameters, Kinetic Analysis, and Operating CostSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Fabrication of rGO/Cuprous Oxide Nanocomposites For Gas SensingDokument6 SeitenFabrication of rGO/Cuprous Oxide Nanocomposites For Gas SensingSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- J Cep 2020 107996Dokument26 SeitenJ Cep 2020 107996SHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Removal of Arsenic From Aqueous Media Using Zeolite Chitosan Nanocomposite Membrane PDFDokument8 SeitenRemoval of Arsenic From Aqueous Media Using Zeolite Chitosan Nanocomposite Membrane PDFSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- J Scitotenv 2019 136364Dokument9 SeitenJ Scitotenv 2019 136364SHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- J Matpr 2021 01 569Dokument5 SeitenJ Matpr 2021 01 569SHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Sensores de NPs Cu2ODokument15 SeitenSensores de NPs Cu2OSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Formacion de Las Nanoparticulas de CU2O-CUODokument14 SeitenFormacion de Las Nanoparticulas de CU2O-CUOSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Paper Labo 3.1Dokument7 SeitenPaper Labo 3.1SHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Aplicaciones de Nanoparticulas de CUODokument10 SeitenAplicaciones de Nanoparticulas de CUOSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Scientific Committee On Consumer Safety SCCS: Opinion On P-AminophenolDokument57 SeitenScientific Committee On Consumer Safety SCCS: Opinion On P-AminophenolSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Ferritaa SurfactantesDokument13 SeitenFerritaa SurfactantesSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Grafeno SintesisDokument5 SeitenGrafeno SintesisSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Design of Experiment For Optimization of Nitrophenol Reduction by Green Synthesized Silver NanocatalystDokument11 SeitenDesign of Experiment For Optimization of Nitrophenol Reduction by Green Synthesized Silver NanocatalystSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Accepted Manuscript: Inorganic Chemistry CommunicationsDokument20 SeitenAccepted Manuscript: Inorganic Chemistry CommunicationsSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Cobalamin Coenzyme Forms Are Not Likely To Be SupeDokument10 SeitenCobalamin Coenzyme Forms Are Not Likely To Be SupeSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Zinc and Magnesium Content of Alkaline Phosphatase From: Escherichia ColiDokument8 SeitenZinc and Magnesium Content of Alkaline Phosphatase From: Escherichia ColiSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Coordination Chemistry Reviews: Karolina Wieszczycka, Katarzyna StaszakDokument12 SeitenCoordination Chemistry Reviews: Karolina Wieszczycka, Katarzyna StaszakSHERLY KIMBERLY RAMOS JESUSNoch keine Bewertungen
- Unit 8Dokument12 SeitenUnit 8georgeclaymensNoch keine Bewertungen
- 11 05 13 Chemistry Electrochemistry Assignment 2Dokument8 Seiten11 05 13 Chemistry Electrochemistry Assignment 2Gadde Gopala KrishnaNoch keine Bewertungen
- Modern Aspects of Electrochemistry No. 18 - Ralph E. WhiteDokument374 SeitenModern Aspects of Electrochemistry No. 18 - Ralph E. WhiteMohammad Usman EhsanNoch keine Bewertungen
- Conducting and Evaluating Galvanic Corrosion Tests in ElectrolytesDokument5 SeitenConducting and Evaluating Galvanic Corrosion Tests in Electrolytesvuqar0979Noch keine Bewertungen
- Bachelor of Technology: Computer Science and EngineeringDokument77 SeitenBachelor of Technology: Computer Science and Engineeringkeerthi vemireddyNoch keine Bewertungen
- AMMO-QT U-Rust SentinelDokument4 SeitenAMMO-QT U-Rust SentinelafernandezmingoNoch keine Bewertungen
- Chemical Changes and Reaction WorksheetDokument2 SeitenChemical Changes and Reaction WorksheetriddhiNoch keine Bewertungen
- 2006 Cheng Etal ElecCommDokument6 Seiten2006 Cheng Etal ElecCommAmri SetyaNoch keine Bewertungen
- Chapter 1928 Electrochemistry 29Dokument76 SeitenChapter 1928 Electrochemistry 29Kent NguyenNoch keine Bewertungen
- A1A EC SENSORS AN2 Design of Electronics For EC Sensors V4Dokument7 SeitenA1A EC SENSORS AN2 Design of Electronics For EC Sensors V4Muhammad RohfadliNoch keine Bewertungen
- O2 Sensors Pi 9049119 en GBDokument6 SeitenO2 Sensors Pi 9049119 en GBمعاذ شوقي العبسيNoch keine Bewertungen
- 11.prinsip Anode Tumbal RevDokument21 Seiten11.prinsip Anode Tumbal RevWahyu WahyurachmatdhaniNoch keine Bewertungen
- The Effect of Pyrite Particle Size On The Electrochemical Dissolution of Gold During CyanidationDokument9 SeitenThe Effect of Pyrite Particle Size On The Electrochemical Dissolution of Gold During CyanidationChristy Alexandra Solano GavelánNoch keine Bewertungen
- New Era Public School: Holiday Homework (2017-18) Class: XII Subject: EnglishDokument64 SeitenNew Era Public School: Holiday Homework (2017-18) Class: XII Subject: EnglishHarvir SinghNoch keine Bewertungen
- Project Bank - Second Semester 2023-24Dokument41 SeitenProject Bank - Second Semester 2023-24SOHINI KAYALNoch keine Bewertungen
- 2-Basic Concepts of Corrosion Science and EngineeringDokument31 Seiten2-Basic Concepts of Corrosion Science and EngineeringMarwin G CrispinoNoch keine Bewertungen
- 10 1016@j Mtcomm 2020 101691Dokument64 Seiten10 1016@j Mtcomm 2020 101691Saran LakshmiNoch keine Bewertungen
- Why Study Fuel CellsDokument10 SeitenWhy Study Fuel CellsSiddhesh BhavsarNoch keine Bewertungen
- Philips 32pfl5606-Fonte+schematic PDFDokument2 SeitenPhilips 32pfl5606-Fonte+schematic PDFGilberto Grandini BitencourtNoch keine Bewertungen
- For Examiner's Use A B16 B17 C18 C19 C20Dokument8 SeitenFor Examiner's Use A B16 B17 C18 C19 C20Muhd FaiZNoch keine Bewertungen
- Vyacheslav F., Panov, Et Al. Torsion Fields and Experiments. Journal of New Energy, Vol 2, No 3-4 (Fall-Winter, 1997)Dokument11 SeitenVyacheslav F., Panov, Et Al. Torsion Fields and Experiments. Journal of New Energy, Vol 2, No 3-4 (Fall-Winter, 1997)Petrut ValentinNoch keine Bewertungen
- Corrosion: Galvanic Corrosion: DefinitionDokument4 SeitenCorrosion: Galvanic Corrosion: Definitionspamalstublieft1832Noch keine Bewertungen
- The "Golden Penny" DemonstrationDokument3 SeitenThe "Golden Penny" DemonstrationOren RosenfeldNoch keine Bewertungen
- Self-Assessments 7 PDFDokument6 SeitenSelf-Assessments 7 PDFUmme Abdullah0% (1)
- Potentiostat: User ManualDokument28 SeitenPotentiostat: User ManualKate AnagnostouNoch keine Bewertungen
- Research ProposalDokument24 SeitenResearch ProposalAisyah SarjuniNoch keine Bewertungen
- (23007591 - Fatigue of Aircraft Structures) Aircraft Corrosion - Review of Corrosion Processes and Its Effects in Selected Cases PDFDokument16 Seiten(23007591 - Fatigue of Aircraft Structures) Aircraft Corrosion - Review of Corrosion Processes and Its Effects in Selected Cases PDFRamsés RamirezNoch keine Bewertungen
- List of Chemistry Mnemonics - WikipediaDokument12 SeitenList of Chemistry Mnemonics - WikipediaSreedevi Krishnakumar0% (1)