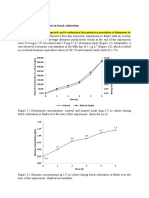
Das könnte Ihnen auch gefallen
- Grupos8 8949 2794331 Tarea1 ProceFrutasVegetalesDokument3 SeitenGrupos8 8949 2794331 Tarea1 ProceFrutasVegetalesAntonio MoncayoNoch keine Bewertungen
- BPE35306 - Microalgae Biotechnology - Practical Manual 2020 - OnlineDokument16 SeitenBPE35306 - Microalgae Biotechnology - Practical Manual 2020 - OnlineAntonio MoncayoNoch keine Bewertungen
- Grupos4 4158 2794143 FrutasyvegetalesDokument4 SeitenGrupos4 4158 2794143 FrutasyvegetalesAntonio MoncayoNoch keine Bewertungen
- Chlorella Repeated Batch Growth Algaemist Panel PBR Day-Night Cycle - 1 - MinDokument110 SeitenChlorella Repeated Batch Growth Algaemist Panel PBR Day-Night Cycle - 1 - MinAntonio MoncayoNoch keine Bewertungen
- Grupos4 - 4158 - 2794142 - Frutas y VerdurasDokument2 SeitenGrupos4 - 4158 - 2794142 - Frutas y VerdurasAntonio MoncayoNoch keine Bewertungen
- Mass and Energy Balances - Background InformationDokument12 SeitenMass and Energy Balances - Background InformationAntonio MoncayoNoch keine Bewertungen
- FPE20306-2019 - Chapter 3 - Evaporation - TheoryDokument18 SeitenFPE20306-2019 - Chapter 3 - Evaporation - TheoryAntonio MoncayoNoch keine Bewertungen
- Exercise 0.4 Dried Potato SlicesDokument25 SeitenExercise 0.4 Dried Potato SlicesAntonio MoncayoNoch keine Bewertungen
- Formula Sheet Mass and Energy BalancesDokument3 SeitenFormula Sheet Mass and Energy BalancesAntonio MoncayoNoch keine Bewertungen
- Picochlorum Repeated Batch Growth Algaemist Panel PBR Day-Night Cycle - 1 - MinDokument110 SeitenPicochlorum Repeated Batch Growth Algaemist Panel PBR Day-Night Cycle - 1 - MinAntonio MoncayoNoch keine Bewertungen
- FPE20306-2019 - Chapter 1 - Food Material Properties - TheoryDokument17 SeitenFPE20306-2019 - Chapter 1 - Food Material Properties - TheoryAntonio MoncayoNoch keine Bewertungen
- Exercise 0.5 UltrafiltrationDokument67 SeitenExercise 0.5 UltrafiltrationAntonio MoncayoNoch keine Bewertungen
- Workassignment BPE IA Tjalma FinalDokument4 SeitenWorkassignment BPE IA Tjalma FinalAntonio MoncayoNoch keine Bewertungen
- 1.1 Effect of Ingredients and Processing Parameters On Pellet QualityDokument8 Seiten1.1 Effect of Ingredients and Processing Parameters On Pellet QualityAntonio MoncayoNoch keine Bewertungen
- Exercise 0.2 Centrifuging Whole MilkDokument32 SeitenExercise 0.2 Centrifuging Whole MilkAntonio MoncayoNoch keine Bewertungen
- Exercise 0.3 Evaporation of Fresh Orange JuiceDokument16 SeitenExercise 0.3 Evaporation of Fresh Orange JuiceAntonio MoncayoNoch keine Bewertungen
- Exercise 5.3 - Spray Dryer I (2020)Dokument36 SeitenExercise 5.3 - Spray Dryer I (2020)Antonio MoncayoNoch keine Bewertungen
- 2020 Superheated Steam Drying v1Dokument1 Seite2020 Superheated Steam Drying v1Antonio MoncayoNoch keine Bewertungen
- FPE30806 Exam Excel File 2020Dokument18 SeitenFPE30806 Exam Excel File 2020Antonio MoncayoNoch keine Bewertungen
- Composition Reduction Dry Milling CornDokument10 SeitenComposition Reduction Dry Milling CornAntonio MoncayoNoch keine Bewertungen
- 0.2 Thermal Characterization and Phase Beahvior of CornstarchDokument7 Seiten0.2 Thermal Characterization and Phase Beahvior of CornstarchAntonio MoncayoNoch keine Bewertungen
- Evaluation Hammer Mill MaizeDokument25 SeitenEvaluation Hammer Mill MaizeAntonio MoncayoNoch keine Bewertungen
- Reflection Report - AMDokument2 SeitenReflection Report - AMAntonio MoncayoNoch keine Bewertungen
- Rafael Ejemplos 1Dokument10 SeitenRafael Ejemplos 1Antonio MoncayoNoch keine Bewertungen
- Results Draft 1 Antonio Moncayo - RCDokument11 SeitenResults Draft 1 Antonio Moncayo - RCAntonio MoncayoNoch keine Bewertungen
- Work Assignment BPE Antonio Moncayo v2Dokument3 SeitenWork Assignment BPE Antonio Moncayo v2Antonio MoncayoNoch keine Bewertungen
- Work assignment-thesisBPE-Wimar R v02Dokument3 SeitenWork assignment-thesisBPE-Wimar R v02Antonio MoncayoNoch keine Bewertungen
- Rafael EjemplosDokument7 SeitenRafael EjemplosAntonio MoncayoNoch keine Bewertungen
- Rafael 3Dokument7 SeitenRafael 3Antonio MoncayoNoch keine Bewertungen
- WA - Ana Sofia Camacho FinalDokument3 SeitenWA - Ana Sofia Camacho FinalAntonio MoncayoNoch keine Bewertungen
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Evidence That Food Proteins in Vaccines Cause The Development of Food Allergies and Its Implications For Vaccine Policy 2329 6631 1000137Dokument3 SeitenEvidence That Food Proteins in Vaccines Cause The Development of Food Allergies and Its Implications For Vaccine Policy 2329 6631 1000137Engkiong GoNoch keine Bewertungen
- Antigen and Its PropertiesDokument20 SeitenAntigen and Its Propertiestusharpremin92% (12)
- Playbook: Use The Resource Links Below With The PDF DownloadDokument20 SeitenPlaybook: Use The Resource Links Below With The PDF DownloadFlo Bors100% (2)
- Seasonal Flu Vaccinations Don't 'Stick' Long-Term in Bone Marrow: Contrast To CHDokument6 SeitenSeasonal Flu Vaccinations Don't 'Stick' Long-Term in Bone Marrow: Contrast To CHz zzzkzmmzNoch keine Bewertungen
- Product Range PDFDokument84 SeitenProduct Range PDFshubhamkatna100% (1)
- Vaccines Research PaperDokument16 SeitenVaccines Research PaperHana Pung100% (1)
- The Safety of Influenza Vaccines in Children An Institute For Vaccine Safety White PaperDokument67 SeitenThe Safety of Influenza Vaccines in Children An Institute For Vaccine Safety White Papermaheshmuralinair6Noch keine Bewertungen
- Veterinary Question and AnswersDokument26 SeitenVeterinary Question and AnswersMuhammad Tufail Ahmed Khan100% (1)
- Novavax Initiates COVID-19 Vaccine Clinical Trial CrossoverDokument2 SeitenNovavax Initiates COVID-19 Vaccine Clinical Trial CrossoverDavid SantosNoch keine Bewertungen
- Adjuvants Used in VaccinesDokument15 SeitenAdjuvants Used in VaccinesVon Valentine MhuteNoch keine Bewertungen
- Immunostimulants Types and FunctionsDokument7 SeitenImmunostimulants Types and FunctionsTanmay MehtaNoch keine Bewertungen
- Antibodies To Squalene in Recipients of Anthrax VaccineDokument9 SeitenAntibodies To Squalene in Recipients of Anthrax VaccineThorsteinn ThorsteinssonNoch keine Bewertungen
- New Generation Vaccines Seminar19Dokument4 SeitenNew Generation Vaccines Seminar19Nerma0% (1)
- WHO IVB 14.07 EngDokument14 SeitenWHO IVB 14.07 EnggineNoch keine Bewertungen
- 130 Studies Linking Vaccines To Neurological and Autoimmune Issues Common To Autism PDFDokument158 Seiten130 Studies Linking Vaccines To Neurological and Autoimmune Issues Common To Autism PDFYonathan Rangel (Phoenix)Noch keine Bewertungen
- Chapter 3Dokument5 SeitenChapter 3Mišel VuittonNoch keine Bewertungen
- Covid-19 Mass Vaccination PlanDokument95 SeitenCovid-19 Mass Vaccination PlanJeremy Turley100% (1)
- Presentation On: Therapeutic Value of AdjuvantsDokument13 SeitenPresentation On: Therapeutic Value of AdjuvantsShamim MahmudNoch keine Bewertungen
- 107 Studies On VaccineDokument65 Seiten107 Studies On VaccineiRepair Gold Coast100% (1)
- 3637 Technical Bulletin MTD Isa 760 vg4536718630311562122Dokument1 Seite3637 Technical Bulletin MTD Isa 760 vg4536718630311562122Thảo ThảoNoch keine Bewertungen
- P3118.1.08.E.1.Coglavax SPCDokument5 SeitenP3118.1.08.E.1.Coglavax SPCJelena Terzic100% (1)
- Dds Report Chapter 16Dokument51 SeitenDds Report Chapter 16Rye M. BirungNoch keine Bewertungen
- Annex 3 Who Trs 962-3Dokument28 SeitenAnnex 3 Who Trs 962-3Miguel Angel CastroNoch keine Bewertungen
- Adjuvants - IntroductionDokument1 SeiteAdjuvants - Introductionsaranya INoch keine Bewertungen
- CORVELVA Metagenomic Analysis Report On Gardasil 9Dokument9 SeitenCORVELVA Metagenomic Analysis Report On Gardasil 9Gene XeneNoch keine Bewertungen
- Sars-Cov-2 RBD Neutralizing Antibody Induction Is Enhanced by Particulate VaccinationDokument11 SeitenSars-Cov-2 RBD Neutralizing Antibody Induction Is Enhanced by Particulate VaccinationmwdhtirahNoch keine Bewertungen
- Amy W's Final Vaccine DocumentDokument59 SeitenAmy W's Final Vaccine DocumentamyNoch keine Bewertungen
- MODULE 2: Types of Vaccine and Adverse ReactionsDokument29 SeitenMODULE 2: Types of Vaccine and Adverse ReactionsnandaNoch keine Bewertungen
- Injectable Biochip For Sars-CoV-2 DetectionDokument8 SeitenInjectable Biochip For Sars-CoV-2 Detectionvojkan73Noch keine Bewertungen
- Part III, Endotoxin Test Concerns of BiologicsDokument7 SeitenPart III, Endotoxin Test Concerns of BiologicsEverton MonteiroNoch keine Bewertungen