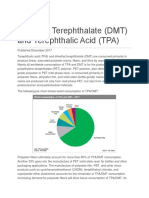
Das könnte Ihnen auch gefallen
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- The FMCG Packaging Moving The Market Towards Sustainable Packaging MaterialsDokument6 SeitenThe FMCG Packaging Moving The Market Towards Sustainable Packaging MaterialsRajib ChatterjeeNoch keine Bewertungen
- Rev JERR 105430 Sya ADokument14 SeitenRev JERR 105430 Sya ALei LopezNoch keine Bewertungen
- Latest Inidutry DatabaseDokument62 SeitenLatest Inidutry DatabaseAbdul RahmanNoch keine Bewertungen
- Plastic Has A Problem Is Chemical Recycling The SolutionDokument12 SeitenPlastic Has A Problem Is Chemical Recycling The SolutionTomGaliciaNoch keine Bewertungen
- About The Use of Recycled or Biodegradable Filaments For Sustainability of 3D PrintingDokument11 SeitenAbout The Use of Recycled or Biodegradable Filaments For Sustainability of 3D PrintingAchyut TrivediNoch keine Bewertungen
- Polymer Processing-2Dokument62 SeitenPolymer Processing-2andrijanto123Noch keine Bewertungen
- GreenDelta-Bottle-Tutorial 1.10 June2020Dokument51 SeitenGreenDelta-Bottle-Tutorial 1.10 June2020Consorcio Integral Ingenieros AmbientalesNoch keine Bewertungen
- Cavite State University: WWW - Cvsu.edu - PHDokument4 SeitenCavite State University: WWW - Cvsu.edu - PHsunshine apuraNoch keine Bewertungen
- Utilization of Waste Plastic in Manufacturing of Paver BlocksDokument4 SeitenUtilization of Waste Plastic in Manufacturing of Paver BlocksAragorn RingsNoch keine Bewertungen
- PolymerDokument6 SeitenPolymerABHAY VISHWAKARMA100% (1)
- Question Regarding Types of PlasticsDokument2 SeitenQuestion Regarding Types of PlasticsFadhlul HadiNoch keine Bewertungen
- Crastin PBT - Rynite Pet PDFDokument38 SeitenCrastin PBT - Rynite Pet PDFkfaravNoch keine Bewertungen
- Materials 14 01162Dokument16 SeitenMaterials 14 01162Mihai Edward GheorgheNoch keine Bewertungen
- Swot Analysis Examples: To Help You Write Your OwnDokument25 SeitenSwot Analysis Examples: To Help You Write Your OwnKassandra KayNoch keine Bewertungen
- Himmelsbach 2020Dokument11 SeitenHimmelsbach 2020Sri AmshaNoch keine Bewertungen
- Lectura 4. Structural Units For Polymers - Osswald (67 - 71)Dokument6 SeitenLectura 4. Structural Units For Polymers - Osswald (67 - 71)Jhon SolanoNoch keine Bewertungen
- Kebutuhan DMT AsiaDokument2 SeitenKebutuhan DMT Asiafebry16pwjNoch keine Bewertungen
- Facts About PET - 25 March 2013Dokument7 SeitenFacts About PET - 25 March 2013Cátia CoelhoNoch keine Bewertungen
- Recycling of Polymers A ReviewDokument15 SeitenRecycling of Polymers A ReviewChristhy Vanessa Ruiz MadroñeroNoch keine Bewertungen
- CE18B037 Assignment2Dokument9 SeitenCE18B037 Assignment2Kota Thapaswini ce18b037Noch keine Bewertungen
- Code of Practice For SuperintendentsDokument74 SeitenCode of Practice For Superintendentsprionus2002Noch keine Bewertungen
- Activation Energy For The Pyrolysis of Polymer WastesDokument6 SeitenActivation Energy For The Pyrolysis of Polymer WastesSwiftTGSolutionsNoch keine Bewertungen
- ISAMP 2021 List Online Presentation ScheduleDokument2 SeitenISAMP 2021 List Online Presentation ScheduleNur DeliveryNoch keine Bewertungen
- Blow MoldingDokument17 SeitenBlow MoldingNilanjana MishraNoch keine Bewertungen
- Esco Waste InceneratorDokument4 SeitenEsco Waste InceneratorRizki Try AtmantiNoch keine Bewertungen
- Introduction To PolymersDokument180 SeitenIntroduction To PolymersDaffa Talitha Widyadhana WidyadhanaNoch keine Bewertungen
- RFP-RVM-Amritsar Smart City PDFDokument54 SeitenRFP-RVM-Amritsar Smart City PDFICT WalaNoch keine Bewertungen
- KKKKKKKKDokument72 SeitenKKKKKKKKkirankalkii44Noch keine Bewertungen
- Polyethylene Terephthalate Waste Recycling and AppDokument10 SeitenPolyethylene Terephthalate Waste Recycling and AppJohn David LunaNoch keine Bewertungen
- Ecoverde BrochureDokument12 SeitenEcoverde BrochureRicardina Rosibel Rodriguez MendozaNoch keine Bewertungen