Das könnte Ihnen auch gefallen
- GEK1532 Color Perception: Thorsten Wohland Dep. of Chemistry S8-03-06 Tel.: 6516 1248 E-Mail: Chmwt@nus - Edu.sgDokument43 SeitenGEK1532 Color Perception: Thorsten Wohland Dep. of Chemistry S8-03-06 Tel.: 6516 1248 E-Mail: Chmwt@nus - Edu.sggivena2ndchanceNoch keine Bewertungen
- GEK1532 PaintersDokument40 SeitenGEK1532 Paintersgivena2ndchanceNoch keine Bewertungen
- GEK1532 ProteinsDokument31 SeitenGEK1532 Proteinsgivena2ndchanceNoch keine Bewertungen
- GEK1532 Light, Spectra and Colour Mixing - 3Dokument49 SeitenGEK1532 Light, Spectra and Colour Mixing - 3givena2ndchanceNoch keine Bewertungen
- GEK1532 Nerve PulsesDokument35 SeitenGEK1532 Nerve Pulsesgivena2ndchanceNoch keine Bewertungen
- GEK1532 Physiology of PerceptionDokument34 SeitenGEK1532 Physiology of Perceptiongivena2ndchanceNoch keine Bewertungen
- GEK1532 Optics of The EyeDokument42 SeitenGEK1532 Optics of The Eyegivena2ndchanceNoch keine Bewertungen
- GEK1532 Interaction of Light With MatterDokument45 SeitenGEK1532 Interaction of Light With Mattergivena2ndchanceNoch keine Bewertungen
- GEK1532 Binocular Vision and Colour LinguisticsDokument44 SeitenGEK1532 Binocular Vision and Colour Linguisticsgivena2ndchanceNoch keine Bewertungen
- GEK1532 Genetics of VisionDokument31 SeitenGEK1532 Genetics of Visiongivena2ndchanceNoch keine Bewertungen
- GEK1532 IntroductionDokument35 SeitenGEK1532 Introductiongivena2ndchanceNoch keine Bewertungen
- GEK1532 Causes of ColourDokument44 SeitenGEK1532 Causes of Colourgivena2ndchanceNoch keine Bewertungen
- GEK1532 Differences in Colour VisionDokument29 SeitenGEK1532 Differences in Colour Visiongivena2ndchanceNoch keine Bewertungen
- GEK1532 History of ColourDokument36 SeitenGEK1532 History of Colourgivena2ndchanceNoch keine Bewertungen
- GEK1532 Color Vision & Binocular VisionDokument43 SeitenGEK1532 Color Vision & Binocular Visiongivena2ndchanceNoch keine Bewertungen
- GEK1045 OthersDokument9 SeitenGEK1045 Othersgivena2ndchanceNoch keine Bewertungen
- GEK1532 Color Classification (CIE)Dokument37 SeitenGEK1532 Color Classification (CIE)givena2ndchanceNoch keine Bewertungen
- GEK1045 EssayDokument10 SeitenGEK1045 Essaygivena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 4Dokument21 SeitenGEK1045 Lecture 4givena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 12 Chinese Religion2Dokument16 SeitenGEK1045 Lecture 12 Chinese Religion2givena2ndchanceNoch keine Bewertungen
- GEK1045 - Intro To World Religion Essay - U090307HDokument7 SeitenGEK1045 - Intro To World Religion Essay - U090307Hgivena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 1Dokument27 SeitenGEK1045 Lecture 1givena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 6Dokument20 SeitenGEK1045 Lecture 6givena2ndchanceNoch keine Bewertungen
- GEK1045 Guidelines For IVLE Forum 0910Dokument5 SeitenGEK1045 Guidelines For IVLE Forum 0910givena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 11 Chinese ReligionDokument18 SeitenGEK1045 Lecture 11 Chinese Religiongivena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 8 HinduismDokument23 SeitenGEK1045 Lecture 8 Hinduismgivena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 5Dokument17 SeitenGEK1045 Lecture 5givena2ndchanceNoch keine Bewertungen
- GEK1045 Hinduism Lecture 7Dokument22 SeitenGEK1045 Hinduism Lecture 7givena2ndchanceNoch keine Bewertungen
- GEK1045 Lecture 2Dokument23 SeitenGEK1045 Lecture 2givena2ndchanceNoch keine Bewertungen
- GEK1045 Essay Questions and Guidelines 0910Dokument4 SeitenGEK1045 Essay Questions and Guidelines 0910givena2ndchanceNoch keine Bewertungen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Novel Alzheimer's Drug Targets Multiple PathwaysDokument4 SeitenNovel Alzheimer's Drug Targets Multiple PathwaysIndra HedarNoch keine Bewertungen
- Abortion II ND Yr MSC NursingDokument121 SeitenAbortion II ND Yr MSC Nursingesthereugenia100% (1)
- Hyperbaric Oxygen Therapy For A Pediatric Electrical BurnDokument3 SeitenHyperbaric Oxygen Therapy For A Pediatric Electrical Burnsarah iriamanaNoch keine Bewertungen
- Mental Health Awareness PowerPointDokument14 SeitenMental Health Awareness PowerPointLoryn Gail BañezNoch keine Bewertungen
- TURP (Transurethral Resection of The Prostate)Dokument5 SeitenTURP (Transurethral Resection of The Prostate)shyam varmaNoch keine Bewertungen
- Label Nama Obat Di Rak ObatDokument8 SeitenLabel Nama Obat Di Rak ObatDyah SariNoch keine Bewertungen
- Comparison Study. CARESCAPEB850Dokument4 SeitenComparison Study. CARESCAPEB850Alberto MHNoch keine Bewertungen
- CryoglobulinsDokument39 SeitenCryoglobulinsanamariavNoch keine Bewertungen
- Definition-Acute Kidney InjuryDokument6 SeitenDefinition-Acute Kidney Injuryashi leginNoch keine Bewertungen
- Cell Injury, Cell Death, and Adaptations: Dr. I Made Naris Pujawan, M.Biomed, SP - PADokument27 SeitenCell Injury, Cell Death, and Adaptations: Dr. I Made Naris Pujawan, M.Biomed, SP - PAlindaNoch keine Bewertungen
- Accurate Diagnosis Is Difficult, Specially in The Latent PhaseDokument4 SeitenAccurate Diagnosis Is Difficult, Specially in The Latent Phasev_vijayakanth7656Noch keine Bewertungen
- Roche Cardiac Reader ManualDokument59 SeitenRoche Cardiac Reader ManualKate Camat FaminialagaoNoch keine Bewertungen
- Reading - Age With Moderate DehydrationDokument8 SeitenReading - Age With Moderate DehydrationSophia IbuyanNoch keine Bewertungen
- Desease Control On GoatsDokument14 SeitenDesease Control On Goats180006 RosmilahNoch keine Bewertungen
- Compilation of Reviewer For Fundamentals of Nursing PDF FreeDokument49 SeitenCompilation of Reviewer For Fundamentals of Nursing PDF FreeTyler VintNoch keine Bewertungen
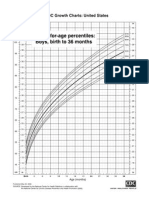
- CDC Boys 0 - 36 Mths - Length-For-AgeDokument1 SeiteCDC Boys 0 - 36 Mths - Length-For-AgepmverlaanNoch keine Bewertungen
- Ocumentation: Analysis of The Situation and Trends in Sunscreen ProductsDokument4 SeitenOcumentation: Analysis of The Situation and Trends in Sunscreen ProductsEvelyn GómezNoch keine Bewertungen
- Fading Rainbow - Abm Sycipgroup 1 1Dokument14 SeitenFading Rainbow - Abm Sycipgroup 1 1Kate ParanaNoch keine Bewertungen
- Asthma and COPDDokument79 SeitenAsthma and COPDDawit g/kidanNoch keine Bewertungen
- Bipolar Disorder Treatment: Psychosocial Therapies Augment Medication to Improve Outcomes and Reduce DisabilityDokument26 SeitenBipolar Disorder Treatment: Psychosocial Therapies Augment Medication to Improve Outcomes and Reduce DisabilityChristine EnriquezNoch keine Bewertungen
- Internet-Related Psychopathology Clinical Phenotypes and PerspectivesDokument138 SeitenInternet-Related Psychopathology Clinical Phenotypes and PerspectivesAndreea NicolaeNoch keine Bewertungen
- Topic 1: © Aspire Training & ConsultingDokument20 SeitenTopic 1: © Aspire Training & ConsultingMashael SulimanNoch keine Bewertungen
- EE MOCK Test 2 Jan 2017 Questions OnlyDokument28 SeitenEE MOCK Test 2 Jan 2017 Questions OnlyEmadSamirNoch keine Bewertungen
- Postural DeviationsDokument40 SeitenPostural DeviationsMohamed Tariq Acchha100% (2)
- Advances in The Treatment of Sickle CellDokument15 SeitenAdvances in The Treatment of Sickle CellMayuri P KNoch keine Bewertungen
- PDF Dis StandardsDokument35 SeitenPDF Dis StandardsEdén PastoraNoch keine Bewertungen
- IEN 420: Environmental & Safety Engineering Spring 2021-2022Dokument34 SeitenIEN 420: Environmental & Safety Engineering Spring 2021-2022Moh HNoch keine Bewertungen
- Endocrinology Take Home ExamDokument7 SeitenEndocrinology Take Home ExamRenz L. SalumbreNoch keine Bewertungen
- Thyroid Disorders in Children and AdolescentsDokument12 SeitenThyroid Disorders in Children and AdolescentsCharity TololiuNoch keine Bewertungen
- Antihuman Globulin TestsDokument6 SeitenAntihuman Globulin TestsElla OrtegaNoch keine Bewertungen