Das könnte Ihnen auch gefallen
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (121)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- Q1 Test3 Test 4Dokument31 SeitenQ1 Test3 Test 4Jennelyn PerezNoch keine Bewertungen
- Nephelometer Onboard GALILEO: "Instrument & Results"Dokument17 SeitenNephelometer Onboard GALILEO: "Instrument & Results"Sanjeev MishraNoch keine Bewertungen
- PH - Buffers 1 QPDokument12 SeitenPH - Buffers 1 QPRoshae SinclairNoch keine Bewertungen
- Cymel-Nf-3030 en A4Dokument1 SeiteCymel-Nf-3030 en A4DidarNoch keine Bewertungen
- Ejemplo DTML Crossflow PDFDokument2 SeitenEjemplo DTML Crossflow PDFDaniel González Juárez100% (1)
- E9) Total Internal ReflectionDokument2 SeitenE9) Total Internal Reflectionlaurencrowe08Noch keine Bewertungen
- Transduction PrinciplesDokument14 SeitenTransduction PrinciplesNavin KaranthNoch keine Bewertungen
- Density and Pressure 2 MSDokument9 SeitenDensity and Pressure 2 MSananafra861Noch keine Bewertungen
- QUESTIONSDokument5 SeitenQUESTIONSkanyakuarNoch keine Bewertungen
- Bes - REDOX TITRATION PDFDokument3 SeitenBes - REDOX TITRATION PDFAvi Thakur100% (1)
- BS 3Dokument58 SeitenBS 3abhishekNoch keine Bewertungen
- Conclusion of The Classical-Nonclassical Ion The Structural of The CationDokument9 SeitenConclusion of The Classical-Nonclassical Ion The Structural of The Cationyonadime922Noch keine Bewertungen
- At Sample Paper 2018 19 C Xii Pass Paper 2Dokument23 SeitenAt Sample Paper 2018 19 C Xii Pass Paper 2Anurag BhattnagarNoch keine Bewertungen
- Problems For Activity 3Dokument2 SeitenProblems For Activity 3richooNoch keine Bewertungen
- Lecture Notes 1 - Fluid MechanicsDokument9 SeitenLecture Notes 1 - Fluid MechanicsJane AndaNoch keine Bewertungen
- Simulacion Aero Enfriador PDFDokument40 SeitenSimulacion Aero Enfriador PDFNicandroGonzalesNoch keine Bewertungen
- ACBPD Lecture4 2017Dokument39 SeitenACBPD Lecture4 2017MohamedTaherNoch keine Bewertungen
- Principles of Organic Synthesis 2th EditionDokument643 SeitenPrinciples of Organic Synthesis 2th Editionminh leNoch keine Bewertungen
- Temperature Regulation During Exercises: Dr. Ghousia ShahidDokument26 SeitenTemperature Regulation During Exercises: Dr. Ghousia ShahidAflaha KhanNoch keine Bewertungen
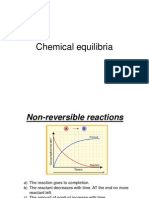
- Chemical EquilibriaDokument15 SeitenChemical EquilibriaiceggNoch keine Bewertungen
- ch8 Crystallization PDFDokument10 Seitench8 Crystallization PDFLakshmi Narayan MeenaNoch keine Bewertungen
- The Mechanism of The Adsorption of Gold Cyanide On Activated CarbonDokument13 SeitenThe Mechanism of The Adsorption of Gold Cyanide On Activated Carbonpakde jongko50% (2)
- Ijertv2is80230 6Dokument13 SeitenIjertv2is80230 6emad hayekNoch keine Bewertungen
- Biochem Lab Act PrelimDokument20 SeitenBiochem Lab Act PrelimcharlesNoch keine Bewertungen
- Square-Wave VoltammetryDokument15 SeitenSquare-Wave VoltammetryNatasa VukicevicNoch keine Bewertungen
- t1-p1-2 Spectra-Structure Correlations in The Mid and Far InfraredDokument34 Seitent1-p1-2 Spectra-Structure Correlations in The Mid and Far InfraredCorvusdav EspinNoch keine Bewertungen
- Review Questions 2024Dokument11 SeitenReview Questions 2024emperial2006Noch keine Bewertungen
- Introduction To Thermofluid MechanicsDokument6 SeitenIntroduction To Thermofluid MechanicsLaki ENNoch keine Bewertungen
- Thermodynamic Properties of Oxygen From 20-100KDokument2 SeitenThermodynamic Properties of Oxygen From 20-100Kwesileh981Noch keine Bewertungen
- Coal GasificationDokument11 SeitenCoal GasificationPratik RanjanNoch keine Bewertungen