Das könnte Ihnen auch gefallen
- HKDSE Chemistry Bridging Programe 1CDokument76 SeitenHKDSE Chemistry Bridging Programe 1Cthe222Noch keine Bewertungen
- Hydrogen ProductiomDokument11 SeitenHydrogen ProductiomKhoirul SaputriNoch keine Bewertungen
- Sulfur and Hydrogen Sulfide RecoveryDokument27 SeitenSulfur and Hydrogen Sulfide RecoveryChemical.AliNoch keine Bewertungen
- Characterisation of Different Commercial Reactive MagnesiaDokument13 SeitenCharacterisation of Different Commercial Reactive MagnesiaSwaroop NarayananNoch keine Bewertungen
- Energy Conversion and ManagementDokument7 SeitenEnergy Conversion and ManagementhusseinhshNoch keine Bewertungen
- Various Methods For Hydrogen Production - After EditDokument11 SeitenVarious Methods For Hydrogen Production - After EditmoftajNoch keine Bewertungen
- Behavior of Sio2 With ResinsDokument10 SeitenBehavior of Sio2 With ResinsPiyush BanerjeeNoch keine Bewertungen
- BiohidrogenDokument11 SeitenBiohidrogenMinea Maria-ramonaNoch keine Bewertungen
- Cement Rotary Kiln Questions & AnswersDokument37 SeitenCement Rotary Kiln Questions & AnswersNael95% (19)
- Novel scheme enables production of renewable green dieselDokument13 SeitenNovel scheme enables production of renewable green dieselNadia RizanedewiNoch keine Bewertungen
- A New Power, Methanol, and DME Polygeneration Process Using Integrated Chemical Looping SystemsDokument15 SeitenA New Power, Methanol, and DME Polygeneration Process Using Integrated Chemical Looping SystemsCriveanuNNarcisNoch keine Bewertungen
- Chapter 23 Thermophilic Biohydrogen Production PDFDokument12 SeitenChapter 23 Thermophilic Biohydrogen Production PDFaegosmithNoch keine Bewertungen
- Catalytic Formation of Lactic and Levulinic Acids From Biomass Derived Monosaccarides Through Sn-Beta Formed by ImpregnationDokument16 SeitenCatalytic Formation of Lactic and Levulinic Acids From Biomass Derived Monosaccarides Through Sn-Beta Formed by ImpregnationAhmed EssamNoch keine Bewertungen
- Processes 09 00436 v4Dokument23 SeitenProcesses 09 00436 v4Maheesha GunathungaNoch keine Bewertungen
- Using Waste CO To Increase Ethanol Production From Corn Ethanol Biorefineries: Techno-Economic AnalysisDokument15 SeitenUsing Waste CO To Increase Ethanol Production From Corn Ethanol Biorefineries: Techno-Economic Analysisnileshparmar21Noch keine Bewertungen
- Renewable HydrogenDokument10 SeitenRenewable HydrogenVedavathi ReddyNoch keine Bewertungen
- Green Chemistry: Accepted ManuscriptDokument10 SeitenGreen Chemistry: Accepted ManuscriptSyarif HidayatNoch keine Bewertungen
- Evaluation of Lignocellulosic Biomass Upgrading Routes To Fuels and ChemicalsDokument21 SeitenEvaluation of Lignocellulosic Biomass Upgrading Routes To Fuels and Chemicals김병철Noch keine Bewertungen
- An Integrated Catalytic Approach For The Production of Hydrogen by GlycerolDokument6 SeitenAn Integrated Catalytic Approach For The Production of Hydrogen by GlycerolMahdy HajienayatiNoch keine Bewertungen
- Sorption Enhanced Steam Reforming (SESR) : A Direct Route Towards Efficient Hydrogen Production From Biomass-Derived CompoundsDokument8 SeitenSorption Enhanced Steam Reforming (SESR) : A Direct Route Towards Efficient Hydrogen Production From Biomass-Derived CompoundsserchNoch keine Bewertungen
- PetroleumDokument13 SeitenPetroleumPrashantNoch keine Bewertungen
- Zhang 2015Dokument7 SeitenZhang 2015AishNoch keine Bewertungen
- Chemical Conversion of Biomass To Green ChemicalsDokument32 SeitenChemical Conversion of Biomass To Green ChemicalsMaria Cecille Sarmiento GarciaNoch keine Bewertungen
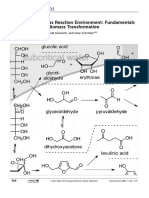
- Subcritical Water As Reaction Environment: Fundamentals of Hydrothermal Biomass TransformationDokument14 SeitenSubcritical Water As Reaction Environment: Fundamentals of Hydrothermal Biomass TransformationSeptian Perwira YudhaNoch keine Bewertungen
- Dnv-Position Paper Co2 Utilization Tcm4-445820Dokument20 SeitenDnv-Position Paper Co2 Utilization Tcm4-445820lhphong021191Noch keine Bewertungen
- Effect of Substrate Concentration On Hydrogen Production andDokument9 SeitenEffect of Substrate Concentration On Hydrogen Production andProfessor Douglas TorresNoch keine Bewertungen
- Simulation Studies On Ethanol Production From Sugar Cane ResiduesDokument22 SeitenSimulation Studies On Ethanol Production From Sugar Cane ResiduesChakuliNoch keine Bewertungen
- 1 s2.0 S0920586109000984 MainDokument11 Seiten1 s2.0 S0920586109000984 MainClaudia Elizabeth Ruiz DávilaNoch keine Bewertungen
- Reator Artigo PDFDokument11 SeitenReator Artigo PDFÊnio BruceNoch keine Bewertungen
- Ferment Able SugarDokument6 SeitenFerment Able SugarXenia MenaNoch keine Bewertungen
- Energies: /Zno/AlDokument25 SeitenEnergies: /Zno/AlAnonymous Ksq1dyPRhNoch keine Bewertungen
- Energies 13 00610 v2 PDFDokument25 SeitenEnergies 13 00610 v2 PDFIsmail ŞahbazNoch keine Bewertungen
- ManuscriptDokument18 SeitenManuscriptNguyen TrangNoch keine Bewertungen
- Kinetics and Reaction Engineering of Levulinic Acid Production From Aqueous Glucose SolutionsDokument12 SeitenKinetics and Reaction Engineering of Levulinic Acid Production From Aqueous Glucose Solutionsראול אפונטהNoch keine Bewertungen
- Development of An Efficient Methanol Production PRDokument31 SeitenDevelopment of An Efficient Methanol Production PRKorean Drama TVNoch keine Bewertungen
- Methanol Production by CO Hydrogenation: Analysis and Simulation of Reactor PerformanceDokument19 SeitenMethanol Production by CO Hydrogenation: Analysis and Simulation of Reactor PerformancehelloNoch keine Bewertungen
- Role of Photocatalysis - Recent AdvancementsDokument20 SeitenRole of Photocatalysis - Recent AdvancementsGRagaNoch keine Bewertungen
- Hydrogen Production From Glycerol An UpdateDokument5 SeitenHydrogen Production From Glycerol An Updateemi_v11Noch keine Bewertungen
- s12649-018-0267-0Dokument13 Seitens12649-018-0267-0Celic RamosNoch keine Bewertungen
- Producing Hydrogen Through Electrolysis and Other ProcessesDokument50 SeitenProducing Hydrogen Through Electrolysis and Other ProcessesAnedo 1909Noch keine Bewertungen
- Journal of CO Utilization: Nora Meiri, Roman Radus, Moti HerskowitzDokument6 SeitenJournal of CO Utilization: Nora Meiri, Roman Radus, Moti HerskowitzFarah Talib Al-sudaniNoch keine Bewertungen
- High PressureDokument20 SeitenHigh PressureJam imtiazNoch keine Bewertungen
- Python Project SynopsisDokument30 SeitenPython Project SynopsisAshish RoshanNoch keine Bewertungen
- Ismaila2021 Article ThermodynamicAnalysisOfSteamReDokument12 SeitenIsmaila2021 Article ThermodynamicAnalysisOfSteamRechairunnisaNoch keine Bewertungen
- Anaerobic Fermentation Hydrogen Production Organic WasteDokument11 SeitenAnaerobic Fermentation Hydrogen Production Organic WasteJesús Alfonso Vázquez BarragánNoch keine Bewertungen
- Carbon Capture: Converting CO2 to Jet Fuel Using Iron CatalystDokument2 SeitenCarbon Capture: Converting CO2 to Jet Fuel Using Iron CatalystWilliamNoch keine Bewertungen
- Solid Acid Catalyzed Aldol Dimerization of Levulinic Acid For The Preparation of C10 Renewable Fuel and Chemical FeedstocksDokument6 SeitenSolid Acid Catalyzed Aldol Dimerization of Levulinic Acid For The Preparation of C10 Renewable Fuel and Chemical FeedstocksLucas de MeloNoch keine Bewertungen
- s2.0 S0920586122X001141 s2.0 S0920586122000992main - PDFX Amz Security Token IQoJbDokument18 Seitens2.0 S0920586122X001141 s2.0 S0920586122000992main - PDFX Amz Security Token IQoJbMasroorAbroNoch keine Bewertungen
- Biogas Purification Using AmineDokument9 SeitenBiogas Purification Using AmineAlbertIvanoAndreanNoch keine Bewertungen
- Preproof HKirschDokument51 SeitenPreproof HKirschMohammed GhanemNoch keine Bewertungen
- Analysis of Operational Issues in Hydrothermal Liquefaction and Supercritical Water Gasification Processes A ReviewDokument28 SeitenAnalysis of Operational Issues in Hydrothermal Liquefaction and Supercritical Water Gasification Processes A ReviewNhean FierceghastNoch keine Bewertungen
- Echno Economic Analysis For ProductionofLArabitol fromLArabinoseDokument8 SeitenEchno Economic Analysis For ProductionofLArabitol fromLArabinoseADITI AWASTHINoch keine Bewertungen
- Materials 16 00472 v2Dokument15 SeitenMaterials 16 00472 v2Claudio Contreras DíazNoch keine Bewertungen
- Production of Renewable Hydrogen by Aqueous-Phase Reforming of Glycerol OverDokument7 SeitenProduction of Renewable Hydrogen by Aqueous-Phase Reforming of Glycerol OverMahdy HajienayatiNoch keine Bewertungen
- ECM - Campanario - Gutierrez - 2017 - FT Biofuels From SCWR - ECM - PreprintDokument40 SeitenECM - Campanario - Gutierrez - 2017 - FT Biofuels From SCWR - ECM - PreprintAlbert StratingNoch keine Bewertungen
- Chemical Engineering Journal: Yunfei He, Luxin Zhang, Yuting Liu, Simin Yi, Han Yu, Yujie Zhu, Ruijun SunDokument12 SeitenChemical Engineering Journal: Yunfei He, Luxin Zhang, Yuting Liu, Simin Yi, Han Yu, Yujie Zhu, Ruijun Sunbruno barrosNoch keine Bewertungen
- Energies: Bio-Crude Production Through Aqueous Phase Recycling of Hydrothermal Liquefaction of Sewage SludgeDokument18 SeitenEnergies: Bio-Crude Production Through Aqueous Phase Recycling of Hydrothermal Liquefaction of Sewage SludgekrisNoch keine Bewertungen
- Selective Hydrolysis of Cellulose and Polysaccharides Into Sugars by Catalytic Hydrothermal Method Using Sulfonated Activated-CarbonDokument14 SeitenSelective Hydrolysis of Cellulose and Polysaccharides Into Sugars by Catalytic Hydrothermal Method Using Sulfonated Activated-CarbonFitri HardiantiNoch keine Bewertungen
- 3-D Multi-Tubular Reactor Model Development For The Oxidative Dehydrogenation of Butene To 1,3-ButadieneDokument21 Seiten3-D Multi-Tubular Reactor Model Development For The Oxidative Dehydrogenation of Butene To 1,3-ButadieneKhabib FirmansyahNoch keine Bewertungen
- New Horizons For Hydrogen: Producing Hydrogen From Renewable ResourcesDokument8 SeitenNew Horizons For Hydrogen: Producing Hydrogen From Renewable ResourcesBraulio BrunaudNoch keine Bewertungen
- Energies: Fractionation For Biodiesel Purification Using Supercritical Carbon DioxideDokument10 SeitenEnergies: Fractionation For Biodiesel Purification Using Supercritical Carbon DioxideArdila Hayu TiwikramaNoch keine Bewertungen
- Nanoporous Catalysts for Biomass ConversionVon EverandNanoporous Catalysts for Biomass ConversionFeng-Shou XiaoNoch keine Bewertungen
- Advances in Biofeedstocks and Biofuels, Volume 2: Production Technologies for BiofuelsVon EverandAdvances in Biofeedstocks and Biofuels, Volume 2: Production Technologies for BiofuelsLalit Kumar SinghNoch keine Bewertungen
- Clean Energy Catalyst: Green Hydrogen Revolutionizing RefiningVon EverandClean Energy Catalyst: Green Hydrogen Revolutionizing RefiningNoch keine Bewertungen
- Rates of Dissolution of Magnesite and Dolomite in EAF SlagsDokument14 SeitenRates of Dissolution of Magnesite and Dolomite in EAF SlagsAgustine SetiawanNoch keine Bewertungen
- 3 5 1Dokument268 Seiten3 5 1rajrudrapaaNoch keine Bewertungen
- Chapter 5 Gastrointestinal Agents Reviewer PDFDokument6 SeitenChapter 5 Gastrointestinal Agents Reviewer PDFdavenNoch keine Bewertungen
- Sip Group 4 Chapter 1Dokument6 SeitenSip Group 4 Chapter 1markism RNNoch keine Bewertungen
- MgO-C Refractories Chapter Focuses on Corrosion ResistanceDokument6 SeitenMgO-C Refractories Chapter Focuses on Corrosion Resistancempaka felliNoch keine Bewertungen
- Chemistry Chapter 2Dokument33 SeitenChemistry Chapter 2Hanzla MangrioNoch keine Bewertungen
- Recycling Spent Magnesia Refractories with Zirconia AdditionsDokument12 SeitenRecycling Spent Magnesia Refractories with Zirconia AdditionsKhaled BOUALINoch keine Bewertungen
- Beltsios Etal 2013 PDFDokument23 SeitenBeltsios Etal 2013 PDFFvg Fvg FvgNoch keine Bewertungen
- S Block-1Dokument46 SeitenS Block-1Jeevan KumarNoch keine Bewertungen
- Is 4737Dokument18 SeitenIs 4737Milagros WieczorekNoch keine Bewertungen
- Is 4041 2006Dokument19 SeitenIs 4041 2006Khan ShahzebNoch keine Bewertungen
- Artal Catalogue PDFDokument72 SeitenArtal Catalogue PDFArtalNoch keine Bewertungen
- Wadi Cement Work Ordinary Portland Cement 53 Grade: Chemical CharacteristicsDokument1 SeiteWadi Cement Work Ordinary Portland Cement 53 Grade: Chemical CharacteristicsAshish SontakkeNoch keine Bewertungen
- 2 - Basic Bricks - Dec 2017 Incl RED2Dokument62 Seiten2 - Basic Bricks - Dec 2017 Incl RED2quỳnh lêNoch keine Bewertungen
- Tos Sem 3 2Dokument46 SeitenTos Sem 3 2Ameya ThanawalaNoch keine Bewertungen
- Refractories - Ceramic Materials That Withstand High TemperaturesDokument11 SeitenRefractories - Ceramic Materials That Withstand High Temperaturesanjali sharmaNoch keine Bewertungen
- Phase Composition of Bauxite-Based Refractory CastablesDokument8 SeitenPhase Composition of Bauxite-Based Refractory CastablesDick ManNoch keine Bewertungen
- FINAL Advanced Materials - COTRONICS - Ceramic AdhesiveDokument15 SeitenFINAL Advanced Materials - COTRONICS - Ceramic AdhesiveBharathidasan SugumaranNoch keine Bewertungen
- ASTM Grades For Insulating Fire BrickDokument2 SeitenASTM Grades For Insulating Fire BrickrajachemNoch keine Bewertungen
- Answer 1:: (Chapter 1) (Chemical Reactions and Equations)Dokument2 SeitenAnswer 1:: (Chapter 1) (Chemical Reactions and Equations)apfc epfoNoch keine Bewertungen
- Kimani - Thermal Conductivity of Refractory Brick Materials PDFDokument200 SeitenKimani - Thermal Conductivity of Refractory Brick Materials PDFelizer eleccionNoch keine Bewertungen
- OPTIMIZING FUEL ADDITIVEDokument12 SeitenOPTIMIZING FUEL ADDITIVEfajar noviantoNoch keine Bewertungen
- Synthesis of Porous Hierarchical Mgo and Its Superb Adsorption PropertiesDokument8 SeitenSynthesis of Porous Hierarchical Mgo and Its Superb Adsorption PropertiesJuan Diego CarrilloNoch keine Bewertungen
- BCC 49 4 2017 PDFDokument276 SeitenBCC 49 4 2017 PDFMariana TavlievaNoch keine Bewertungen
- Cambridge IGCSE: Combined Science 0653/32Dokument24 SeitenCambridge IGCSE: Combined Science 0653/32annwong85Noch keine Bewertungen
- Assessment of Blast Furnace Behaviour Through Softening-Melting TestDokument10 SeitenAssessment of Blast Furnace Behaviour Through Softening-Melting TestvidhyasagarNoch keine Bewertungen