Das könnte Ihnen auch gefallen
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Molecular Dynamics Simulations: Che 520 Fall 2009Dokument8 SeitenMolecular Dynamics Simulations: Che 520 Fall 2009Fabio OkamotoNoch keine Bewertungen
- 2011 - Molecular Dynamics Study of Particle Impact Induced BondDokument95 Seiten2011 - Molecular Dynamics Study of Particle Impact Induced BondthamaraikannanNoch keine Bewertungen
- Prepared By: THAMARAIKANNAN Requested By: Tuesday, February 20, 2018Dokument16 SeitenPrepared By: THAMARAIKANNAN Requested By: Tuesday, February 20, 2018thamaraikannanNoch keine Bewertungen
- Strength-Of - Materials by SubramaniDokument1.044 SeitenStrength-Of - Materials by SubramaniBarinder SinghNoch keine Bewertungen
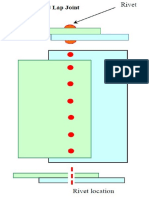
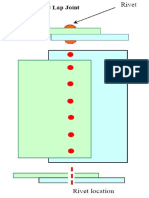
- Lap Joint1 PDFDokument1 SeiteLap Joint1 PDFthamaraikannanNoch keine Bewertungen
- As Lab Manual Dept of Ane: S.No Experiment Page. NoDokument1 SeiteAs Lab Manual Dept of Ane: S.No Experiment Page. NothamaraikannanNoch keine Bewertungen
- Lap JointDokument1 SeiteLap JointthamaraikannanNoch keine Bewertungen
- Lap Joint PDFDokument1 SeiteLap Joint PDFthamaraikannanNoch keine Bewertungen
- 27 Riveted Joints Types and UsesDokument11 Seiten27 Riveted Joints Types and UsesPRASAD326100% (1)
- Lap Joint1Dokument2 SeitenLap Joint1thamaraikannanNoch keine Bewertungen
- 27 Riveted Joints Types and UsesDokument11 Seiten27 Riveted Joints Types and UsesPRASAD326100% (1)
- Ansys ManualDokument47 SeitenAnsys ManualAshwinkumar MallikarjunaNoch keine Bewertungen
- AeroSpace Vechicle Structure IIDokument2 SeitenAeroSpace Vechicle Structure IIthamaraikannanNoch keine Bewertungen
- CAAVD SyllabusDokument1 SeiteCAAVD SyllabusthamaraikannanNoch keine Bewertungen
- Engineering Analysis of Flight Vehicles - SyllabusDokument1 SeiteEngineering Analysis of Flight Vehicles - SyllabusthamaraikannanNoch keine Bewertungen
- Cluster User Guide PDFDokument21 SeitenCluster User Guide PDFthamaraikannanNoch keine Bewertungen
- Aircraft Structures LabDokument84 SeitenAircraft Structures LabthamaraikannanNoch keine Bewertungen
- 2 PAX Short Course Fibers PDFDokument73 Seiten2 PAX Short Course Fibers PDFthamaraikannanNoch keine Bewertungen
- Ase ManualDokument258 SeitenAse ManualthamaraikannanNoch keine Bewertungen
- Claim FormDokument4 SeitenClaim FormthamaraikannanNoch keine Bewertungen
- X - PAX Short Course - Material QualificationDokument18 SeitenX - PAX Short Course - Material QualificationthamaraikannanNoch keine Bewertungen
- Latex For Word Users PDFDokument45 SeitenLatex For Word Users PDFthamaraikannanNoch keine Bewertungen
- Latex For Word Users PDFDokument45 SeitenLatex For Word Users PDFthamaraikannanNoch keine Bewertungen
- Drdo Industry CompendiumDokument578 SeitenDrdo Industry CompendiumthamaraikannanNoch keine Bewertungen
- Calibration of NonlocalDokument7 SeitenCalibration of NonlocalthamaraikannanNoch keine Bewertungen
- Molecular Simulation TechniquesDokument146 SeitenMolecular Simulation Techniquesthamaraikannan100% (1)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (890)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- GPSA Engineering Data Book 14th Edition: Revision Date Reason (S) For RevisionDokument9 SeitenGPSA Engineering Data Book 14th Edition: Revision Date Reason (S) For Revisionrkm_rkmNoch keine Bewertungen
- Semiconductors CBSE Board QuestionsDokument9 SeitenSemiconductors CBSE Board QuestionsnithishmjmmNoch keine Bewertungen
- Martensitic Transformations and Strengthening MechanismsDokument6 SeitenMartensitic Transformations and Strengthening Mechanismsmans2014Noch keine Bewertungen
- Central Angles PDFDokument8 SeitenCentral Angles PDFJoseph LeeNoch keine Bewertungen
- VCE Chemistry Unit 3Dokument484 SeitenVCE Chemistry Unit 3Danny GoldstoneNoch keine Bewertungen
- Classical Electrodynamics Part IIDokument331 SeitenClassical Electrodynamics Part IIPankaj SolankiNoch keine Bewertungen
- Power Transformers: Principles and ApplicationsDokument14 SeitenPower Transformers: Principles and Applicationsvikalp321Noch keine Bewertungen
- MOS Transistor Device OperationDokument15 SeitenMOS Transistor Device OperationSuhas ShirolNoch keine Bewertungen
- Physics For Master PlumbersDokument65 SeitenPhysics For Master PlumbersghkotxcgjfjerrNoch keine Bewertungen
- Models - Mixer.centrifugal PumpDokument22 SeitenModels - Mixer.centrifugal Pumpcarlos tNoch keine Bewertungen
- Review PaperDokument13 SeitenReview PaperKomal SinghNoch keine Bewertungen
- EXAM Rev 1 BiomechanicsDokument12 SeitenEXAM Rev 1 BiomechanicsuhbfouinNoch keine Bewertungen
- Atomic Emission Spectroscopy AsdaDokument18 SeitenAtomic Emission Spectroscopy AsdaMark Cliffton BadlonNoch keine Bewertungen
- Sobretensiones y Coordinacione de AislamientoDokument280 SeitenSobretensiones y Coordinacione de AislamientomartinpellsNoch keine Bewertungen
- Kinetic Theory Explains Gas LawsDokument4 SeitenKinetic Theory Explains Gas LawsAntonique HeadmanNoch keine Bewertungen
- EMCO Compact 5 CNC Instructor ManualDokument331 SeitenEMCO Compact 5 CNC Instructor ManualJames Hyde83% (6)
- Pentode Straped TriodeDokument32 SeitenPentode Straped Triodebigpriap100% (1)
- IRFS4229Dokument9 SeitenIRFS4229EDERTREVISANNoch keine Bewertungen
- m1 Dynamics Connected ParticlesDokument17 Seitenm1 Dynamics Connected ParticlesAjmalMansoorNoch keine Bewertungen
- Hossein Rezvantalab, PH.D.: Professional ProfileDokument5 SeitenHossein Rezvantalab, PH.D.: Professional Profileindebtanup2443Noch keine Bewertungen
- 02 Statics of Rigid Bodies 01 ConceptsDokument3 Seiten02 Statics of Rigid Bodies 01 Conceptsxaaabbb_550464353Noch keine Bewertungen
- Covennatone Line Notation PDFDokument5 SeitenCovennatone Line Notation PDFifiokNoch keine Bewertungen
- OpenFOAM StressDokument11 SeitenOpenFOAM StresssemetayNoch keine Bewertungen
- Orthogonal Vs OrthonormalDokument2 SeitenOrthogonal Vs OrthonormalShouman BaruaNoch keine Bewertungen
- Setting Out Circular CurvesDokument21 SeitenSetting Out Circular CurvesvpmohammedNoch keine Bewertungen
- Exact Solutions Navier Stokes WangDokument19 SeitenExact Solutions Navier Stokes WangToddharrisNoch keine Bewertungen
- Eltjo HaselhoffDokument2 SeitenEltjo HaselhoffJavier Valentin Gonzalez ReynaNoch keine Bewertungen
- U2 - L10 Stress Isobar or Pressure Bulb PDFDokument6 SeitenU2 - L10 Stress Isobar or Pressure Bulb PDFศิวาเวช อบมาNoch keine Bewertungen
- Building Mat 08-09Dokument39 SeitenBuilding Mat 08-09takishiNoch keine Bewertungen