Das könnte Ihnen auch gefallen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- Flux Skimming PDFDokument6 SeitenFlux Skimming PDFpbp2956Noch keine Bewertungen
- Micro Project MRSDokument13 SeitenMicro Project MRS너사랑Noch keine Bewertungen
- G+12 Raft Foundation ReportDokument13 SeitenG+12 Raft Foundation ReportvinujohnpanickerNoch keine Bewertungen
- 2 Parking Garage - Shotcrete - 1002Dokument1 Seite2 Parking Garage - Shotcrete - 1002VJ QatarNoch keine Bewertungen
- Green Building and Sustainable Construction: CE142 Dr. J. Berlin P. JuanzonDokument56 SeitenGreen Building and Sustainable Construction: CE142 Dr. J. Berlin P. JuanzonJoseph Berlin Juanzon100% (1)
- CTM Geosynthetics Uniaxial-UnlockedDokument1 SeiteCTM Geosynthetics Uniaxial-UnlockedGautam RaiNoch keine Bewertungen
- PR-3Dokument1 SeitePR-3Fernanda QuelNoch keine Bewertungen
- JKR 2012 SITE INVESTIGATION SCHEDULE OF RATESDokument0 SeitenJKR 2012 SITE INVESTIGATION SCHEDULE OF RATESAjoy Zulfadhli0% (1)
- PLUMBING AND SANITARY FIXTURES GUIDEDokument53 SeitenPLUMBING AND SANITARY FIXTURES GUIDEKristine Malbas100% (1)
- Thermal Analysis: Terry A. Ring Chemical Engineering University of UtahDokument18 SeitenThermal Analysis: Terry A. Ring Chemical Engineering University of Utahrereru zuharaNoch keine Bewertungen
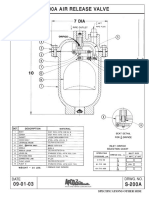
- APCO. S-200a Air Release ValveDokument2 SeitenAPCO. S-200a Air Release ValveAlberto Diaz100% (2)
- 1) Design A Large Building WITHOUT Expansion JointDokument7 Seiten1) Design A Large Building WITHOUT Expansion JointSubinDesarNoch keine Bewertungen
- ECSS E ST 32 02C - Rev.1 (15november2008)Dokument76 SeitenECSS E ST 32 02C - Rev.1 (15november2008)jsadachiNoch keine Bewertungen
- Hot Isostatic Pressing CeramicsDokument4 SeitenHot Isostatic Pressing CeramicsamirsuryahidayahNoch keine Bewertungen
- Corosion Assignment FullDokument104 SeitenCorosion Assignment FullVelavan KNoch keine Bewertungen
- SR20715152848 - Very ImportantDokument6 SeitenSR20715152848 - Very Importantlogan firemanNoch keine Bewertungen
- Waterproofing MethodsDokument21 SeitenWaterproofing MethodsM.IDREES KhanNoch keine Bewertungen
- ASTM STP538 - Cleaning Stainless SteelDokument11 SeitenASTM STP538 - Cleaning Stainless SteelPeter FowlesNoch keine Bewertungen
- iRSVP 3100 Data Sheet.Dokument2 SeiteniRSVP 3100 Data Sheet.Gỗ MộcNoch keine Bewertungen
- Nitoproof 600: Liquid Applied, Elastomeric, One Coat Waterproofing MembraneDokument3 SeitenNitoproof 600: Liquid Applied, Elastomeric, One Coat Waterproofing MembraneFarah HaseenahNoch keine Bewertungen
- 52 Week PPM Planner Template-15 Jul 14Dokument26 Seiten52 Week PPM Planner Template-15 Jul 14safetydellNoch keine Bewertungen
- 2000 IBC Handbook Seismic & WindDokument11 Seiten2000 IBC Handbook Seismic & WindcristinelbNoch keine Bewertungen
- 2099 Robor Corrugated Liet PDFDokument2 Seiten2099 Robor Corrugated Liet PDFJosef StrydomNoch keine Bewertungen
- Princesa Bridge Narrative ReportDokument9 SeitenPrincesa Bridge Narrative ReportRodelo OngNoch keine Bewertungen
- Variable Air Volume SystemDokument62 SeitenVariable Air Volume SystemdokundotNoch keine Bewertungen
- Underground Pipe THK CalDokument12 SeitenUnderground Pipe THK Calmkchy12100% (3)
- CHAPTER 4 - General Models of FoulingDokument10 SeitenCHAPTER 4 - General Models of Foulingkim haksongNoch keine Bewertungen
- Practice problems-STEELDokument14 SeitenPractice problems-STEELreanNoch keine Bewertungen
- مقاومه مواد محاضرات د.فراتDokument121 Seitenمقاومه مواد محاضرات د.فراتlana_salahadinNoch keine Bewertungen
- Dissertation CGDokument147 SeitenDissertation CGChiheb BaNoch keine Bewertungen