Das könnte Ihnen auch gefallen
- Cap 27 Trastornos de La Conduccion y Del Ritmo CardiacoDokument35 SeitenCap 27 Trastornos de La Conduccion y Del Ritmo CardiacoAlejandrafnNoch keine Bewertungen
- Areas de BrodmannDokument7 SeitenAreas de BrodmannLaura Valbuena100% (1)
- Manual de Enfermeria Extrahospitalario 2Dokument257 SeitenManual de Enfermeria Extrahospitalario 2Angel Cotta100% (1)
- CardiotonicosDokument28 SeitenCardiotonicosLourdes Brizuela100% (1)
- HEMOFILIADokument5 SeitenHEMOFILIAJuan Velasquez100% (1)
- TUMOR PARDO ResumenDokument4 SeitenTUMOR PARDO ResumenJorge SolisNoch keine Bewertungen
- Composicion de La Saliva BioquiicaDokument7 SeitenComposicion de La Saliva BioquiicaAmber Williams33% (3)
- Anemia Microcítica Con Caso ClinicoDokument7 SeitenAnemia Microcítica Con Caso ClinicoOscar Cortez HerreraNoch keine Bewertungen
- Biometría Hemática OdontoDokument2 SeitenBiometría Hemática OdontonaddhxielyNoch keine Bewertungen
- FRACTURAS-MAXILARES PacompiaDokument20 SeitenFRACTURAS-MAXILARES PacompiaLizbeth PalliNoch keine Bewertungen
- Micro AlbuminuriaDokument3 SeitenMicro AlbuminuriaJorge OntanedaNoch keine Bewertungen
- Biometría Hemática CompletaDokument5 SeitenBiometría Hemática CompletaSandra EspinosaNoch keine Bewertungen
- Lupus EritematosoDokument6 SeitenLupus EritematosoLucia Lucero Paz OliveraNoch keine Bewertungen
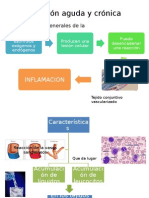
- Inflamacion Aguda y CronicaDokument48 SeitenInflamacion Aguda y CronicaCindy PumaderaNoch keine Bewertungen
- Osteítis DeformanteDokument7 SeitenOsteítis DeformanteAlexandra EANoch keine Bewertungen
- HemofiliaDokument15 SeitenHemofiliaSamantha SanRey100% (1)
- Análisis de Orina ECODokument7 SeitenAnálisis de Orina ECOJonathan LazarinosNoch keine Bewertungen
- Clase 1 Biofilm DentalDokument25 SeitenClase 1 Biofilm DentalJuanPablo FuentesNoch keine Bewertungen
- HemofiliaDokument53 SeitenHemofiliaDaniielita Beltran50% (2)
- Dentífricos y ColutoriosDokument17 SeitenDentífricos y ColutoriosFelipe RoncerosNoch keine Bewertungen
- Condiciones No Patologícas de La Cavidad BucalDokument9 SeitenCondiciones No Patologícas de La Cavidad BucalEduardo GuzamanahNoch keine Bewertungen
- Micro AlbuminuriaDokument4 SeitenMicro Albuminuriamarielasofy100% (1)
- Fisiologia de La SangreDokument123 SeitenFisiologia de La SangreBPMNoch keine Bewertungen
- ANFOTERICINADokument6 SeitenANFOTERICINAEvelyn Jiménez MacharéNoch keine Bewertungen
- A Anomalías Del Desarrollo de CaraDokument13 SeitenA Anomalías Del Desarrollo de CaraDianitaVelezPincayNoch keine Bewertungen
- Introduccion FarmacologiaDokument19 SeitenIntroduccion FarmacologiaLeonardoFlores100% (2)
- Cuadro HematicoDokument38 SeitenCuadro HematicoAna SofiaNoch keine Bewertungen
- Linfoma No HodgkinDokument68 SeitenLinfoma No HodgkinEdithRuelasRodriguezNoch keine Bewertungen
- Quiste de Los MaxilaresDokument5 SeitenQuiste de Los MaxilaresGina IgnacioNoch keine Bewertungen
- Protesis SomaticaDokument12 SeitenProtesis SomaticaxtianBVNoch keine Bewertungen
- Enfermedad de PagetDokument8 SeitenEnfermedad de Pagetapi-3740897100% (1)
- HemofiliaDokument5 SeitenHemofiliaYarela VelardeNoch keine Bewertungen
- FluorosisDokument28 SeitenFluorosisLaura RojasNoch keine Bewertungen
- 6 Fluido GingivalDokument17 Seiten6 Fluido GingivalDana Lisett Ygnacio LlanosNoch keine Bewertungen
- Teoria Placa Ecologica MarshDokument2 SeitenTeoria Placa Ecologica MarshFelipe Hernandez100% (1)
- Candidiasis Oral Por Candida AlbicansDokument19 SeitenCandidiasis Oral Por Candida AlbicansCeses100% (1)
- Papiloma EscamosoDokument3 SeitenPapiloma EscamosoChristi A. NavaNoch keine Bewertungen
- HIPERAMONEMIADokument13 SeitenHIPERAMONEMIADiegoAlejandroCardona100% (1)
- Patologías Infecciosas de Origen Odontolgénico, CirugíaDokument6 SeitenPatologías Infecciosas de Origen Odontolgénico, CirugíaFrancisca SotoNoch keine Bewertungen
- Pruebas de Función HepáticaDokument5 SeitenPruebas de Función Hepáticagian_corbascioNoch keine Bewertungen
- Shock AnafilácticoDokument14 SeitenShock Anafilácticonadinek96Noch keine Bewertungen
- Diapositivas. Sars Cov 2Dokument23 SeitenDiapositivas. Sars Cov 2Samira NamiNoch keine Bewertungen
- Hemostasia y TrombosisDokument43 SeitenHemostasia y TrombosisLuis Mérida de LeónNoch keine Bewertungen
- Conteo de HematiesDokument7 SeitenConteo de HematiesPedro Mengole100% (1)
- Anemia AplásicaDokument51 SeitenAnemia AplásicaElizabeth PalaciosNoch keine Bewertungen
- Linfoma Hodgkin y No HodgkinDokument48 SeitenLinfoma Hodgkin y No HodgkinAndrés Cabrera50% (2)
- Trastornos HemodinamicosDokument5 SeitenTrastornos HemodinamicospatriciaaleivaNoch keine Bewertungen
- Hemoglobina Glicosilada Nuevas PerspectivasDokument9 SeitenHemoglobina Glicosilada Nuevas PerspectivasErick RiveraNoch keine Bewertungen
- Clasificacion de Fracturas Maxilar y MadibulaDokument4 SeitenClasificacion de Fracturas Maxilar y MadibulaAlejandra Del Valle LauNoch keine Bewertungen
- Anestesicos Locales Farmacologia en OdontologiaDokument61 SeitenAnestesicos Locales Farmacologia en OdontologiaMatthew Lama Cordova100% (1)
- Glandulas SalivalesDokument39 SeitenGlandulas SalivalesDIEGO ALONSO LOZANO H.Noch keine Bewertungen
- Clase 2.2 Streptococcus y EnterococcusDokument60 SeitenClase 2.2 Streptococcus y EnterococcusAriel Molina ValenzuelaNoch keine Bewertungen
- Homeostasis Del Calcio)Dokument46 SeitenHomeostasis Del Calcio)Keylyn ZelayaNoch keine Bewertungen
- Antecedentes Históricos Del DerechoDokument7 SeitenAntecedentes Históricos Del DerechoNathanaelNoch keine Bewertungen
- Cáncer OralDokument4 SeitenCáncer OralLaura Giraldo Quintero100% (1)
- Hematofisiología y HemostasiaDokument15 SeitenHematofisiología y HemostasiaJHOKABED KAFIRA RIVERA BARRETONoch keine Bewertungen
- Vibrio CholeraeDokument4 SeitenVibrio CholeraedianacontrerasNoch keine Bewertungen
- Dientes SupernumerariosDokument12 SeitenDientes SupernumerariosCeses100% (1)
- Mecanismos de Lesión CelularDokument7 SeitenMecanismos de Lesión CelularEnrique CorreaNoch keine Bewertungen
- Hemofilia ADokument37 SeitenHemofilia ANilreyam FerNoch keine Bewertungen
- Hemofilia PDFDokument26 SeitenHemofilia PDFMirthyan ZafraNoch keine Bewertungen
- Trastornos de La CoagulacionDokument4 SeitenTrastornos de La CoagulacionMaria Fernandez100% (1)
- Hemo FiliaDokument5 SeitenHemo Filianoxphy.vrthrNoch keine Bewertungen
- Norma de Referencia - PPT Serums MayoDokument31 SeitenNorma de Referencia - PPT Serums MayoDiana De La CruzNoch keine Bewertungen
- Estudios MyC - Virtual - Estudios MyC - Virtu@l - 02 (PARTE A)Dokument10 SeitenEstudios MyC - Virtual - Estudios MyC - Virtu@l - 02 (PARTE A)Diana De La Cruz100% (1)
- APENDICITIS AGUDA Basado en Preguntas PDFDokument32 SeitenAPENDICITIS AGUDA Basado en Preguntas PDFDiana De La Cruz100% (2)
- Presencial Examen N 1 PDFDokument12 SeitenPresencial Examen N 1 PDFDiana De La CruzNoch keine Bewertungen
- Presencial Examen N 1 PDFDokument12 SeitenPresencial Examen N 1 PDFDiana De La CruzNoch keine Bewertungen
- Gin EcoDokument30 SeitenGin EcoOmayra Chincha100% (3)
- Farmacocinetica 2016 PDFDokument15 SeitenFarmacocinetica 2016 PDFDiana De La CruzNoch keine Bewertungen
- Examen Essalud 2010Dokument11 SeitenExamen Essalud 2010Ruben Quispe Villamonte100% (1)
- Red PNIE 2013 MMarzalDokument27 SeitenRed PNIE 2013 MMarzalDiana De La CruzNoch keine Bewertungen
- VIH en PediatríaDokument38 SeitenVIH en PediatríaDiana De La CruzNoch keine Bewertungen
- Alfa FetoproteinaDokument4 SeitenAlfa FetoproteinaQUIMICO CLINICO WILLIANS SANCHEZ100% (3)
- Antibioticos Cofepris Mayo-10Dokument37 SeitenAntibioticos Cofepris Mayo-10Carolina Durán100% (1)
- Uso Racional de AntibioticosDokument28 SeitenUso Racional de AntibioticosareseoNoch keine Bewertungen
- Ges Maria RamirezDokument1 SeiteGes Maria RamirezMARIA ESTERNoch keine Bewertungen
- Nauseas y VomitosDokument34 SeitenNauseas y VomitosAnthonyBautistaParionaNoch keine Bewertungen
- CASO CLINICO - Anaplasma Phagocitophilum en Un Perro PDFDokument4 SeitenCASO CLINICO - Anaplasma Phagocitophilum en Un Perro PDFDianaCalaNoch keine Bewertungen
- Caso Clinico Sindrome NefroticoDokument14 SeitenCaso Clinico Sindrome NefroticoNinde Rivera Gonzalez0% (1)
- VOLVULODokument6 SeitenVOLVULOAndreyIvanovNoch keine Bewertungen
- Taller Auditoria de Gestion Exposiciones Caso PracticoDokument8 SeitenTaller Auditoria de Gestion Exposiciones Caso Practicojulio jaimesNoch keine Bewertungen
- Periodo Pre, Trans y Post OperatorioDokument18 SeitenPeriodo Pre, Trans y Post OperatorioLissette Mendoza100% (1)
- Tarjetas Farmacológ02Dokument46 SeitenTarjetas Farmacológ02Liss Pamee Davila50% (8)
- Poli Gabinte BimDokument31 SeitenPoli Gabinte BimMario Morales SantanaNoch keine Bewertungen
- Homotoxicología en PsiquiatríaDokument5 SeitenHomotoxicología en PsiquiatríaCelia SteimanNoch keine Bewertungen
- Aborto EspontáneoDokument9 SeitenAborto EspontáneoLiceo CegbymNoch keine Bewertungen
- Árboles de Decisión en Enfermedades de TiroidesDokument14 SeitenÁrboles de Decisión en Enfermedades de TiroidesLuis Alberto Tercero Silva100% (2)
- 07capitulo Signos Vitales AlexDokument5 Seiten07capitulo Signos Vitales AlexManu PadillaNoch keine Bewertungen
- 3.esofagitis CausticaDokument30 Seiten3.esofagitis CausticaVanessa HidalgoNoch keine Bewertungen
- ESOFAGELIMINADA9MARZ18Dokument141 SeitenESOFAGELIMINADA9MARZ18Octavio GuerraNoch keine Bewertungen
- Gammagrafia PulmonarDokument9 SeitenGammagrafia PulmonarPablo Andrés RamírezNoch keine Bewertungen
- Composición Química de La MarihuanaDokument4 SeitenComposición Química de La MarihuanaSantiago Vargas Ore100% (1)
- OXIGENOTERAPIA ResumenDokument7 SeitenOXIGENOTERAPIA ResumenMary Cruz C. CNoch keine Bewertungen
- Muestreo de RedesDokument5 SeitenMuestreo de RedesyemiurNoch keine Bewertungen
- Embarazo Alto Bajo RiesgoDokument35 SeitenEmbarazo Alto Bajo RiesgoFrancis DeibisNoch keine Bewertungen
- Endoftalmitis BacterianaDokument25 SeitenEndoftalmitis BacterianaCortesia Guipe VANoch keine Bewertungen
- Callista RoyDokument7 SeitenCallista RoyAntonio González BenítezNoch keine Bewertungen
- Enfermedades EmbarazpDokument93 SeitenEnfermedades EmbarazpFiorella SYNoch keine Bewertungen
- Formulario de Información Profundo para Esencias Florales para Adultos Con Síntomas de Trastorno Por Déficit de Atención e HiperactividadDokument7 SeitenFormulario de Información Profundo para Esencias Florales para Adultos Con Síntomas de Trastorno Por Déficit de Atención e HiperactividadPamela Claudia Cordova SilvaNoch keine Bewertungen