Das könnte Ihnen auch gefallen
- Seguimiento de Proyectos ObjetivosDokument12 SeitenSeguimiento de Proyectos ObjetivosArqcast Solis TrujilloNoch keine Bewertungen
- Kiids Rbank Balanced Fund 2qtr 2016Dokument2 SeitenKiids Rbank Balanced Fund 2qtr 2016Rawlinson TolentinoNoch keine Bewertungen
- De-Bottle Necking Polyethylene Plant Due To Butene Shortage: A Multi-Attribute Decision Analysis Using SmartDokument5 SeitenDe-Bottle Necking Polyethylene Plant Due To Butene Shortage: A Multi-Attribute Decision Analysis Using SmartRawlinson TolentinoNoch keine Bewertungen
- Us 2975201Dokument3 SeitenUs 2975201Rawlinson TolentinoNoch keine Bewertungen
- 14 Haz MaterialsDokument81 Seiten14 Haz MaterialsDarby MorganNoch keine Bewertungen
- De-Bottle Necking Polyethylene Plant Due To Butene Shortage: A Multi-Attribute Decision Analysis Using SmartDokument5 SeitenDe-Bottle Necking Polyethylene Plant Due To Butene Shortage: A Multi-Attribute Decision Analysis Using SmartRawlinson TolentinoNoch keine Bewertungen
- 15338-NT15338 Technical PyrazineDokument29 Seiten15338-NT15338 Technical PyrazineRawlinson TolentinoNoch keine Bewertungen
- United States Patent Of?ce: Patented June 17, 1969Dokument3 SeitenUnited States Patent Of?ce: Patented June 17, 1969Rawlinson TolentinoNoch keine Bewertungen
- Effective PresentationsDokument53 SeitenEffective PresentationsShreesh Chandra100% (1)
- Shell Momentum BalancesDokument48 SeitenShell Momentum BalancesJuan CarvajalNoch keine Bewertungen
- CrackingDokument20 SeitenCrackingNiaz Ali KhanNoch keine Bewertungen
- 10%DP12mos-BF W/ BANK CHARGES Amaia Land Corp. Amaia Scapes LipaDokument1 Seite10%DP12mos-BF W/ BANK CHARGES Amaia Land Corp. Amaia Scapes LipaRawlinson TolentinoNoch keine Bewertungen
- Mws Che Inp TXT Direct ExamplesDokument7 SeitenMws Che Inp TXT Direct ExamplesRawlinson TolentinoNoch keine Bewertungen
- Mws Che Inp TXT Direct ExamplesDokument7 SeitenMws Che Inp TXT Direct ExamplesRawlinson TolentinoNoch keine Bewertungen
- IV. Desgn Eqtn For Lamnar TurbulentDokument45 SeitenIV. Desgn Eqtn For Lamnar TurbulentRawlinson TolentinoNoch keine Bewertungen
- Final Candle Paper PDFDokument14 SeitenFinal Candle Paper PDFRawlinson TolentinoNoch keine Bewertungen
- Drainage From Tank With Cylindrical Cross-Section: Discharge Coefficient 'CD' (Assumed)Dokument6 SeitenDrainage From Tank With Cylindrical Cross-Section: Discharge Coefficient 'CD' (Assumed)Rawlinson TolentinoNoch keine Bewertungen
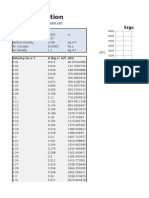
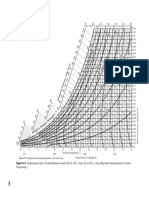
- Ergun EquationDokument4 SeitenErgun EquationRawlinson TolentinoNoch keine Bewertungen
- Centrifugal Pump Curves GdafadasgadDokument2 SeitenCentrifugal Pump Curves GdafadasgadRawlinson TolentinoNoch keine Bewertungen
- Experienced Based Rules of Chemical EngineeringDokument33 SeitenExperienced Based Rules of Chemical Engineeringintania66Noch keine Bewertungen
- Pump Min Safe Flow CalcDokument2 SeitenPump Min Safe Flow CalcmohamedbadawyNoch keine Bewertungen
- Condensate Line SizingDokument2 SeitenCondensate Line Sizingemmanuilmoulos6339100% (1)
- Experienced Based Rules of Chemical EngineeringDokument33 SeitenExperienced Based Rules of Chemical Engineeringintania66Noch keine Bewertungen
- Effective PresentationsDokument45 SeitenEffective PresentationsRawlinson TolentinoNoch keine Bewertungen
- Compressed Air Option CalculatorDokument17 SeitenCompressed Air Option CalculatorEnigmatic Ashutosh KumarNoch keine Bewertungen
- Compressed Air Option CalculatorDokument17 SeitenCompressed Air Option CalculatorEnigmatic Ashutosh KumarNoch keine Bewertungen
- Insulation Thickness Heat Transfer Pipe CalculationsDokument2 SeitenInsulation Thickness Heat Transfer Pipe Calculationsrajpal14667% (6)
- Gas Flow in OrificeDokument2 SeitenGas Flow in OrificeRawlinson TolentinoNoch keine Bewertungen
- Equipment & vessel diagrams guideDokument1 SeiteEquipment & vessel diagrams guideRawlinson TolentinoNoch keine Bewertungen
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeVon EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeBewertung: 4 von 5 Sternen4/5 (5784)
- The Little Book of Hygge: Danish Secrets to Happy LivingVon EverandThe Little Book of Hygge: Danish Secrets to Happy LivingBewertung: 3.5 von 5 Sternen3.5/5 (399)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceVon EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceBewertung: 4 von 5 Sternen4/5 (890)
- Shoe Dog: A Memoir by the Creator of NikeVon EverandShoe Dog: A Memoir by the Creator of NikeBewertung: 4.5 von 5 Sternen4.5/5 (537)
- Grit: The Power of Passion and PerseveranceVon EverandGrit: The Power of Passion and PerseveranceBewertung: 4 von 5 Sternen4/5 (587)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureVon EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureBewertung: 4.5 von 5 Sternen4.5/5 (474)
- The Yellow House: A Memoir (2019 National Book Award Winner)Von EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Bewertung: 4 von 5 Sternen4/5 (98)
- Team of Rivals: The Political Genius of Abraham LincolnVon EverandTeam of Rivals: The Political Genius of Abraham LincolnBewertung: 4.5 von 5 Sternen4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItVon EverandNever Split the Difference: Negotiating As If Your Life Depended On ItBewertung: 4.5 von 5 Sternen4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerVon EverandThe Emperor of All Maladies: A Biography of CancerBewertung: 4.5 von 5 Sternen4.5/5 (271)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryVon EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryBewertung: 3.5 von 5 Sternen3.5/5 (231)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaVon EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaBewertung: 4.5 von 5 Sternen4.5/5 (265)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersVon EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersBewertung: 4.5 von 5 Sternen4.5/5 (344)
- On Fire: The (Burning) Case for a Green New DealVon EverandOn Fire: The (Burning) Case for a Green New DealBewertung: 4 von 5 Sternen4/5 (72)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyVon EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyBewertung: 3.5 von 5 Sternen3.5/5 (2219)
- The Unwinding: An Inner History of the New AmericaVon EverandThe Unwinding: An Inner History of the New AmericaBewertung: 4 von 5 Sternen4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreVon EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreBewertung: 4 von 5 Sternen4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)Von EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Bewertung: 4.5 von 5 Sternen4.5/5 (119)
- Her Body and Other Parties: StoriesVon EverandHer Body and Other Parties: StoriesBewertung: 4 von 5 Sternen4/5 (821)
- 3.1 The Truth About Air TravelDokument14 Seiten3.1 The Truth About Air TravelСвітлана Свирид0% (1)
- Material Safety Data Sheet - MSDS: Section 1. Chemical Product and Company IdentificationDokument5 SeitenMaterial Safety Data Sheet - MSDS: Section 1. Chemical Product and Company IdentificationPubcrawlNoch keine Bewertungen
- Coley A4Dokument49 SeitenColey A4mfiarkeeaNoch keine Bewertungen
- Overview of Pathophysiology of Hypoxemia and HypoxiaDokument15 SeitenOverview of Pathophysiology of Hypoxemia and HypoxiaMARY ANN CAGATANNoch keine Bewertungen
- Australian 9 Grade Physics Lesson 1Dokument32 SeitenAustralian 9 Grade Physics Lesson 1binoyrajcrNoch keine Bewertungen
- Applied Acoustics: André M.N. Spillere, Augusto A. Medeiros, Julio A. CordioliDokument13 SeitenApplied Acoustics: André M.N. Spillere, Augusto A. Medeiros, Julio A. CordioliAbdelali MoumenNoch keine Bewertungen
- Data Biostataplus Sbi2014-EDokument4 SeitenData Biostataplus Sbi2014-ELucila Milagros PinillosNoch keine Bewertungen
- Assignment #1: 1 HgjyygbykvrfDokument1 SeiteAssignment #1: 1 HgjyygbykvrfJuan Sebastian ArangoNoch keine Bewertungen
- 1B Cosmos-Standard - Technical - Guide - v40Dokument45 Seiten1B Cosmos-Standard - Technical - Guide - v40carla deiddaNoch keine Bewertungen
- Tween 80Dokument11 SeitenTween 80fvdxrgNoch keine Bewertungen
- CEFIC Guidelines Transport Equipment Packed Cargo (2010)Dokument7 SeitenCEFIC Guidelines Transport Equipment Packed Cargo (2010)sl1828Noch keine Bewertungen
- Paradise Pools Flyer With Price ListDokument5 SeitenParadise Pools Flyer With Price ListKhuzaima HussainNoch keine Bewertungen
- Tramadol Drug StudyDokument1 SeiteTramadol Drug Studymilkv82% (11)
- S10 Electric Power PackDokument12 SeitenS10 Electric Power PackrolandNoch keine Bewertungen
- Dcom QuestionDokument3 SeitenDcom Questionsushant sahooNoch keine Bewertungen
- Horizontal Projectile WSDokument3 SeitenHorizontal Projectile WSForsbergPhysicsNoch keine Bewertungen
- Hydraulic Power Steering System Design PDFDokument16 SeitenHydraulic Power Steering System Design PDFAdrianBirsan100% (1)
- MATERIAL SAFETY DATA SHEET FOR PREVENTOL-D6 PRESERVATIVEDokument3 SeitenMATERIAL SAFETY DATA SHEET FOR PREVENTOL-D6 PRESERVATIVEAkshay PushpanNoch keine Bewertungen
- CORRELATION AND LINEAR REGRESSIONDokument9 SeitenCORRELATION AND LINEAR REGRESSIONSANKET GANDHINoch keine Bewertungen
- Chapter 1 - Introduction To Machinery PrinciplesDokument27 SeitenChapter 1 - Introduction To Machinery PrinciplesYousab CreatorNoch keine Bewertungen
- 2009 ESC Guidelines On EndocarditisDokument45 Seiten2009 ESC Guidelines On EndocarditisDaondy Friarsa SoehartoNoch keine Bewertungen
- Protreat Hydro EngrgDokument6 SeitenProtreat Hydro EngrgAmitkumar SinghNoch keine Bewertungen
- Home Contents Vehicle Boat Cover Policy Sample Westpac NZDokument27 SeitenHome Contents Vehicle Boat Cover Policy Sample Westpac NZRobin Rutter-BaumannNoch keine Bewertungen
- Operating and Installation Guide For The Digital Instrument: Motoscope Tiny / Speedster / VintageDokument12 SeitenOperating and Installation Guide For The Digital Instrument: Motoscope Tiny / Speedster / Vintagepeter timmermansNoch keine Bewertungen
- Service Manual: EQ1030T47D-820 Light Commercial TruckDokument175 SeitenService Manual: EQ1030T47D-820 Light Commercial TruckYonny ColqueNoch keine Bewertungen
- Lab Journal 4 14032023 104921amDokument8 SeitenLab Journal 4 14032023 104921amHammad MashwaniNoch keine Bewertungen
- Carta Psicrometrica PDFDokument2 SeitenCarta Psicrometrica PDFJuliethNoch keine Bewertungen
- English Test 6Dokument87 SeitenEnglish Test 6Ha PhanNoch keine Bewertungen
- 1 s2.0 S2214860417301148 Main PDFDokument16 Seiten1 s2.0 S2214860417301148 Main PDFQuy Hoang KimNoch keine Bewertungen
- ASME B31.4-2016 Pipeline Transportation Systems For Liquids and SlurriesDokument1 SeiteASME B31.4-2016 Pipeline Transportation Systems For Liquids and SlurriesJose Rodrigo Salguero DuranNoch keine Bewertungen