Das könnte Ihnen auch gefallen
- Crystallography and Mineral Classification GuideDokument6 SeitenCrystallography and Mineral Classification GuideJaniel Arlan OmboyNoch keine Bewertungen
- Catalog Current Update Eye CoverDokument14 SeitenCatalog Current Update Eye CoverTiago MüllerNoch keine Bewertungen
- Where did That Number Come From?: Chronological Histories and Derivations of Numbers Important in ScienceVon EverandWhere did That Number Come From?: Chronological Histories and Derivations of Numbers Important in ScienceNoch keine Bewertungen
- CrystallographyDokument25 SeitenCrystallographyAkhil Sai KrishnaNoch keine Bewertungen
- Kepler's Laws Lab: Ellipses, Speeds, PeriodsDokument15 SeitenKepler's Laws Lab: Ellipses, Speeds, PeriodsArsalan RiazNoch keine Bewertungen
- July 11 Eclipse Myths and Legends Coming AliveDokument4 SeitenJuly 11 Eclipse Myths and Legends Coming AliveJerry VanoNoch keine Bewertungen
- Discover a public domain book scanned by GoogleDokument170 SeitenDiscover a public domain book scanned by GoogleJohn BestNoch keine Bewertungen
- 01-Crystal Orientation ManualDokument76 Seiten01-Crystal Orientation Manualmemento50% (2)
- The Key to Life on Earth: How the Tilt of the Earth's Axis Defines Seasons and Squares the CircleDokument21 SeitenThe Key to Life on Earth: How the Tilt of the Earth's Axis Defines Seasons and Squares the CircleSatyanneshi ERNoch keine Bewertungen
- PolyhedronDokument15 SeitenPolyhedronIIRemmyII100% (1)
- A Market Approach To Forecasting: Background, Theory and PracticeDokument220 SeitenA Market Approach To Forecasting: Background, Theory and PracticeGeorge TziralisNoch keine Bewertungen
- ASTM002 The Galaxy Course Notes 2006 (QMUL)Dokument128 SeitenASTM002 The Galaxy Course Notes 2006 (QMUL)ucaptd3Noch keine Bewertungen
- Thesis Text - Part 1Dokument360 SeitenThesis Text - Part 1Hmt NmslNoch keine Bewertungen
- Colour Centers in Crystals PDFDokument2 SeitenColour Centers in Crystals PDFEricaNoch keine Bewertungen
- 21 SimilarityBetweenQuantumMechanicsAstrologyDokument7 Seiten21 SimilarityBetweenQuantumMechanicsAstrologySudhir Kumar PandeyNoch keine Bewertungen
- Compatibility AstrologyDokument2 SeitenCompatibility AstrologyAlise NiNoch keine Bewertungen
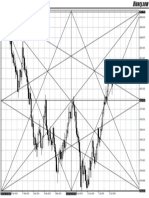
- Gann Grid - Research Work - Volatility Index 10 - DailyDokument1 SeiteGann Grid - Research Work - Volatility Index 10 - DailyWilson NwaiwuNoch keine Bewertungen
- Penicka - Men of Faith - Stravinsky MaritainDokument41 SeitenPenicka - Men of Faith - Stravinsky MaritainMatteo MacinantiNoch keine Bewertungen
- Signs of The ZodiacDokument272 SeitenSigns of The Zodiacashinobioshi0% (1)
- Astrosophic Principles - J Hazelrigg (1917)Dokument135 SeitenAstrosophic Principles - J Hazelrigg (1917)KoayeNoch keine Bewertungen
- Kepler’s Nested Platonic Solids TheoryDokument6 SeitenKepler’s Nested Platonic Solids TheoryMarius Swart100% (1)
- Fine-Structure Constant From Golden Ratio GeometryDokument17 SeitenFine-Structure Constant From Golden Ratio GeometryMichael A. SherbonNoch keine Bewertungen
- Unit 2 SSDokument11 SeitenUnit 2 SShuylimalaNoch keine Bewertungen
- Madelung constant calculationDokument22 SeitenMadelung constant calculationSatyajit biswas100% (1)
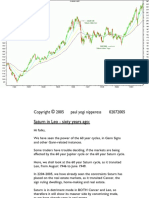
- Djia LeoDokument3 SeitenDjia LeoturtlespiritflutesNoch keine Bewertungen
- Magic Numbers, Planetary Tones and The Body The Evolution of Daoist Inner Alchemy Into Modern Sacred Science (Michael Winn) (Z-Library)Dokument83 SeitenMagic Numbers, Planetary Tones and The Body The Evolution of Daoist Inner Alchemy Into Modern Sacred Science (Michael Winn) (Z-Library)Prometheus AlberioneNoch keine Bewertungen
- Crystallography and Crystal StructuresDokument60 SeitenCrystallography and Crystal Structuresstewarthillclark100% (1)
- Heavenly SignsDokument2 SeitenHeavenly SignsJerry VanoNoch keine Bewertungen
- Theory of RelativityDokument242 SeitenTheory of RelativityMichael RichardiNoch keine Bewertungen
- Golden Ratio Sequence of Rational NumbersDokument25 SeitenGolden Ratio Sequence of Rational NumbersSadi66550% (1)
- A Simple Explicit Model: Short Term Planetary OrbitsDokument7 SeitenA Simple Explicit Model: Short Term Planetary Orbitsbrown0Noch keine Bewertungen
- Crystal Palace Festival Whats On GuideDokument9 SeitenCrystal Palace Festival Whats On GuideGunpowder StudiosNoch keine Bewertungen
- 14 Spiral GalaxiesDokument19 Seiten14 Spiral GalaxiesSam PicassoNoch keine Bewertungen
- Garnet: Garnets (Dokument14 SeitenGarnet: Garnets (Ausu100% (1)
- Measuring The Stars (Chps. 16-17)Dokument5 SeitenMeasuring The Stars (Chps. 16-17)Yoyo Kirby2Noch keine Bewertungen
- Mineral Physics Crystallography A Handbook of Physical Constants9780875908526Dokument359 SeitenMineral Physics Crystallography A Handbook of Physical Constants9780875908526geopicNoch keine Bewertungen
- Uk Rock Identification GuideDokument52 SeitenUk Rock Identification GuideVICTOR MANUEL URIBE CORDOVANoch keine Bewertungen
- Christen-S Astro Position Paper UAC 2012Dokument19 SeitenChristen-S Astro Position Paper UAC 2012Anonymous b2k9ABe7eNoch keine Bewertungen
- Crystallography and Mineralogy NotesDokument220 SeitenCrystallography and Mineralogy NotesSyed Aquib ShamshadNoch keine Bewertungen
- SFAS Dispositor TreesDokument28 SeitenSFAS Dispositor TreesdonkeykongukNoch keine Bewertungen
- Planets and Periodic TableDokument9 SeitenPlanets and Periodic TableDADI PRASANNANoch keine Bewertungen
- Michael Harney and Ioannis Iraklis Haranas - The Dark Energy ProblemDokument3 SeitenMichael Harney and Ioannis Iraklis Haranas - The Dark Energy ProblemMuths999999Noch keine Bewertungen
- Side Real Time-Basics2Dokument30 SeitenSide Real Time-Basics2dombipinNoch keine Bewertungen
- Garnet Nomenclature 2013Dokument55 SeitenGarnet Nomenclature 2013Eric BernardNoch keine Bewertungen
- H.K. Moffatt - Cosmic Dynamos: From Alpha To OmegaDokument5 SeitenH.K. Moffatt - Cosmic Dynamos: From Alpha To OmegaVortices3443Noch keine Bewertungen
- Zodiac: For The East Asian Zodiac Based On The Jovian Orbital Cycle, See - For Other Uses, SeeDokument5 SeitenZodiac: For The East Asian Zodiac Based On The Jovian Orbital Cycle, See - For Other Uses, SeemackNoch keine Bewertungen
- What Is The Solunar TheoryDokument5 SeitenWhat Is The Solunar Theoryobx4ever100% (1)
- Tri State Minerals A Basic GuideDokument7 SeitenTri State Minerals A Basic GuideChristopher WisemanNoch keine Bewertungen
- Graham Yewbrey - John Dee and The Sidney Group Cosmopolitics and Pro cd3 Id1366077362 Size30196Dokument469 SeitenGraham Yewbrey - John Dee and The Sidney Group Cosmopolitics and Pro cd3 Id1366077362 Size30196bluemonarch11100% (1)
- An Introduction To Crystallography and Mineral Crystal SystemsDokument30 SeitenAn Introduction To Crystallography and Mineral Crystal SystemsLuis Paez80% (5)
- Measuring cosmic distances with Cepheid variables and Type Ia supernovaeDokument8 SeitenMeasuring cosmic distances with Cepheid variables and Type Ia supernovaeUSER111Noch keine Bewertungen
- Richard Amoroso - Time$ - 6р PDFDokument6 SeitenRichard Amoroso - Time$ - 6р PDFАнатолий ПодругинNoch keine Bewertungen
- Binary Stars: Dr. H. S. Das Department of Physics Assam University, SilcharDokument19 SeitenBinary Stars: Dr. H. S. Das Department of Physics Assam University, SilcharHimadri Sekhar DasNoch keine Bewertungen
- SU99Dokument95 SeitenSU99saopauloNoch keine Bewertungen
- Liquid Crystals: Properties, Phases and UsesDokument15 SeitenLiquid Crystals: Properties, Phases and UsesReddyvari VenugopalNoch keine Bewertungen
- Crystallography Exercise on Cuprite StructureDokument12 SeitenCrystallography Exercise on Cuprite StructureJuan Javier Marrugo Hernandez0% (1)
- The Planet of Love and The Two PillarsDokument22 SeitenThe Planet of Love and The Two PillarsKim Graae MunchNoch keine Bewertungen
- Dr. Goulden's Astrology Foundations Reading ListDokument1 SeiteDr. Goulden's Astrology Foundations Reading Listcannizzo45091Noch keine Bewertungen
- Avoid Costly Materials mistakes-CEDokument23 SeitenAvoid Costly Materials mistakes-CEManish542Noch keine Bewertungen
- Daftar Lazim Obat Dewasa Dan Anak, Bayi: AntibiotikDokument8 SeitenDaftar Lazim Obat Dewasa Dan Anak, Bayi: AntibiotikkunkunNoch keine Bewertungen
- About The Dyes: Dyes For Cellulose FibersDokument4 SeitenAbout The Dyes: Dyes For Cellulose FibersmanqabatNoch keine Bewertungen
- Chemistry Jun 2010 Mark Scheme Unit 4Dokument22 SeitenChemistry Jun 2010 Mark Scheme Unit 4dylandonNoch keine Bewertungen
- S&T Roofing Solutions Product Guide Mar2018-DP1 0Dokument12 SeitenS&T Roofing Solutions Product Guide Mar2018-DP1 0sattar12345Noch keine Bewertungen
- Cathodic ProtectionDokument132 SeitenCathodic Protectionpeyman_tNoch keine Bewertungen
- Water Pollution Problems and SolutionsDokument56 SeitenWater Pollution Problems and Solutionsarief muhammadNoch keine Bewertungen
- 3. Materials and MethodsDokument42 Seiten3. Materials and MethodsAli Akand AsifNoch keine Bewertungen
- Pipes, Tubes, Fittings, FlangesDokument121 SeitenPipes, Tubes, Fittings, FlangesCarl Jones100% (2)
- LIST OF TSD FACILITIESDokument18 SeitenLIST OF TSD FACILITIESAmelia SantosNoch keine Bewertungen
- 0-5303 Opt PDFDokument226 Seiten0-5303 Opt PDFAnonymous wUv02fNoch keine Bewertungen
- The Fundamental Building Block: The CellDokument12 SeitenThe Fundamental Building Block: The CellTanish JenaNoch keine Bewertungen
- Toxicology: Metal Chosen: MERCURYDokument13 SeitenToxicology: Metal Chosen: MERCURYsamarpita senguptaNoch keine Bewertungen
- Purpose of Master Batch PPA in PEDokument49 SeitenPurpose of Master Batch PPA in PEin_abhay2706Noch keine Bewertungen
- MATERIAL TAKE-OFF LIST FOR SAUDI ARABCO GAS PROJECTDokument12 SeitenMATERIAL TAKE-OFF LIST FOR SAUDI ARABCO GAS PROJECTrajum465Noch keine Bewertungen
- Msds FFFP 3% UlDokument4 SeitenMsds FFFP 3% UlOtto JamesNoch keine Bewertungen
- Soundsafe Safety Data Sheet EnglishDokument5 SeitenSoundsafe Safety Data Sheet Englishard127Noch keine Bewertungen
- Test Report: Noida Testing Laboratories, Noida G T - 20, SECTOR - 117, NOIDA, G. B. NAGAR, U.P., 201 316Dokument1 SeiteTest Report: Noida Testing Laboratories, Noida G T - 20, SECTOR - 117, NOIDA, G. B. NAGAR, U.P., 201 316Kushal Sharma0% (1)
- Designs CatalogDokument77 SeitenDesigns CatalogGen MendozaNoch keine Bewertungen
- Fire cable performance standardsDokument3 SeitenFire cable performance standardsRajan Varghese100% (1)
- API Standard 614 - Lubrication, Shaft-Sealing, and Control-Oil Systems For Special-Purpose ApplicationDokument4 SeitenAPI Standard 614 - Lubrication, Shaft-Sealing, and Control-Oil Systems For Special-Purpose ApplicationFabioSalaNoch keine Bewertungen
- Cap TradeDokument8 SeitenCap TradeEkopribadiNoch keine Bewertungen
- 3656-3756 ML Brochure GouldsDokument44 Seiten3656-3756 ML Brochure Gouldsjulio.esc7Noch keine Bewertungen
- Nhat Huy FillerDokument7 SeitenNhat Huy FillerLê NhungNoch keine Bewertungen
- Lecture 2 - Cellulose Structure PDFDokument55 SeitenLecture 2 - Cellulose Structure PDFpipers10Noch keine Bewertungen
- Antimony Deposit Types & Origins: The Composite Gold-Antimony Vein Deposit at Kharma (Bolivia)Dokument2 SeitenAntimony Deposit Types & Origins: The Composite Gold-Antimony Vein Deposit at Kharma (Bolivia)Milton ObandoNoch keine Bewertungen
- A4-80 Stainless Steel Grade Specification - Midland Bright SteelsDokument4 SeitenA4-80 Stainless Steel Grade Specification - Midland Bright SteelsramonaghergheNoch keine Bewertungen
- 6+7fertilisation Low Cost DripDokument21 Seiten6+7fertilisation Low Cost DripArnab MondalNoch keine Bewertungen
- Understanding and Managing Cell Culture Contamination PDFDokument24 SeitenUnderstanding and Managing Cell Culture Contamination PDFPabloski AndreNoch keine Bewertungen
- METAL FABRICATION RAW MATERIALS GUIDEDokument8 SeitenMETAL FABRICATION RAW MATERIALS GUIDEEdbert TulipasNoch keine Bewertungen