Das könnte Ihnen auch gefallen
- Jack Westin MCAT Content General ChemistryDokument25 SeitenJack Westin MCAT Content General ChemistryLora100% (1)
- Oil & Gas Industry SurveyDokument33 SeitenOil & Gas Industry SurveyRuben PauwelsNoch keine Bewertungen
- Homework 2 SolutionDokument9 SeitenHomework 2 Solutionmohammed el erianNoch keine Bewertungen
- Why Are Cells So Small MyDokument2 SeitenWhy Are Cells So Small Myapi-521781723Noch keine Bewertungen
- G11 Phy CH-8 Study MaterialDokument29 SeitenG11 Phy CH-8 Study MaterialPrasanna VijayakumarNoch keine Bewertungen
- Lecture 15Dokument22 SeitenLecture 15Alim MustofaNoch keine Bewertungen
- Non Newtonian FluidDokument31 SeitenNon Newtonian FluidUzumaki28Noch keine Bewertungen
- Fluid Non NewtonianDokument31 SeitenFluid Non NewtonianUzumaki28Noch keine Bewertungen
- 앳킨스 물리화학 10판 기말범위 - 솔루션Dokument21 Seiten앳킨스 물리화학 10판 기말범위 - 솔루션한상호Noch keine Bewertungen
- No NewtDokument27 SeitenNo NewtLeonardo Escobar GómezNoch keine Bewertungen
- SOL. - P2 - XII Adv. PCM - 23.08.2022Dokument22 SeitenSOL. - P2 - XII Adv. PCM - 23.08.2022BetaNoch keine Bewertungen
- Physical Chemistry Complete Outlines 2017Dokument20 SeitenPhysical Chemistry Complete Outlines 2017Aicha DahmaniNoch keine Bewertungen
- Mass Transfer To Suspensions of Small ParticlesDokument13 SeitenMass Transfer To Suspensions of Small ParticlesrushdiNoch keine Bewertungen
- Chapter 2Dokument25 SeitenChapter 2Sayyed Ali MirjafariNoch keine Bewertungen
- Lab Iv. Silicon Diode Characteristics: 1. ObjectiveDokument9 SeitenLab Iv. Silicon Diode Characteristics: 1. ObjectiveRaja MariyappanNoch keine Bewertungen
- 3 1-LaserintroDokument9 Seiten3 1-LaserintrofvakhacbmjmaqdnrzhNoch keine Bewertungen
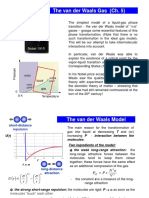
- Lecture 15. The Van Der Waals Gas (Ch. 5) : Nobel 1910Dokument22 SeitenLecture 15. The Van Der Waals Gas (Ch. 5) : Nobel 1910Ihab El SawiNoch keine Bewertungen
- Chap 6 Viscous Fluid in Closed ConduitsDokument40 SeitenChap 6 Viscous Fluid in Closed ConduitsNg Guan ShengNoch keine Bewertungen
- CH3 - Properties of Gases and Vapours-2Dokument18 SeitenCH3 - Properties of Gases and Vapours-2abdoasdafm7Noch keine Bewertungen
- CHM 211Dokument19 SeitenCHM 211Necherem MissionNoch keine Bewertungen
- PaulI 1 Basics of Vacuum TechnologyDokument30 SeitenPaulI 1 Basics of Vacuum TechnologyBill KroulisNoch keine Bewertungen
- DILL CH - 14 Phase EquilibriumDokument18 SeitenDILL CH - 14 Phase EquilibriumMohamed MahmoudKhattabNoch keine Bewertungen
- Jee Mainpapersol2018 PDFDokument22 SeitenJee Mainpapersol2018 PDFBhoopesh KayapakNoch keine Bewertungen
- Fluidmechanics MercrediDokument24 SeitenFluidmechanics MercrediNadiaa AdjoviNoch keine Bewertungen
- Pendahuluan Fisika Zat PadatDokument18 SeitenPendahuluan Fisika Zat PadatMeritania YusmanNoch keine Bewertungen
- Formula ListDokument3 SeitenFormula ListRaihanNoch keine Bewertungen
- ConductivityDokument40 SeitenConductivityhkaruvathilNoch keine Bewertungen
- Lab4 F15 Si DiodeDokument15 SeitenLab4 F15 Si DiodeJohn MarkNoch keine Bewertungen
- Cy 2013Dokument14 SeitenCy 2013pramodNoch keine Bewertungen
- Modern Physics-02-Objective Solved Problems1Dokument5 SeitenModern Physics-02-Objective Solved Problems1Raju SinghNoch keine Bewertungen
- Chemistry Final Step-C Solutions - Gaseous StateDokument6 SeitenChemistry Final Step-C Solutions - Gaseous StateAnas KhalidNoch keine Bewertungen
- Daily Tutorial Sheet 2 JEE Main (Archive) : Solution - Workbook-1 21 Atomic StructureDokument3 SeitenDaily Tutorial Sheet 2 JEE Main (Archive) : Solution - Workbook-1 21 Atomic StructureHalfborn GundersonNoch keine Bewertungen
- Lect1 1Dokument17 SeitenLect1 1Борис ПолякNoch keine Bewertungen
- Lecture07 P2Dokument17 SeitenLecture07 P2Trân TerryNoch keine Bewertungen
- Luid Tatics and Urface Ension: 4-2 Surface Tension and Contact Angle 4-1 Fluid StaticsDokument10 SeitenLuid Tatics and Urface Ension: 4-2 Surface Tension and Contact Angle 4-1 Fluid Statics丁偉庭Noch keine Bewertungen
- The Arge Adron Ollider: Andreas Morsch CERN Physics DivisionDokument46 SeitenThe Arge Adron Ollider: Andreas Morsch CERN Physics DivisionMayank VachherNoch keine Bewertungen
- Atoms and Molecules, Even Uncharged Ones, Are Attracted To Each OtherDokument19 SeitenAtoms and Molecules, Even Uncharged Ones, Are Attracted To Each OtherMohamed MahmoudKhattabNoch keine Bewertungen
- Chap3 FundamentalsInviscidDokument45 SeitenChap3 FundamentalsInviscidSolar Boat TwenteNoch keine Bewertungen
- Bernoulli Equation Part 1Dokument34 SeitenBernoulli Equation Part 1Ashish RanjanNoch keine Bewertungen
- Chem 16 2nd Long Exam Reviewer 2 (Answer Key)Dokument2 SeitenChem 16 2nd Long Exam Reviewer 2 (Answer Key)ben_aldaveNoch keine Bewertungen
- Phychem Jul 6,2018Dokument25 SeitenPhychem Jul 6,2018jantskieNoch keine Bewertungen
- Chemistryb AbehDokument17 SeitenChemistryb AbehAlok BhoiNoch keine Bewertungen
- Carbon Nanotube Field-Effect Transistors and Their Possible ApplicationsDokument36 SeitenCarbon Nanotube Field-Effect Transistors and Their Possible ApplicationsAkshay PatharkarNoch keine Bewertungen
- Newtonian and Non Newtonian FluidsDokument24 SeitenNewtonian and Non Newtonian FluidsatchutNoch keine Bewertungen
- 3.B2. SUPP - Sackur-Tetrode EquationDokument15 Seiten3.B2. SUPP - Sackur-Tetrode EquationHarishNoch keine Bewertungen
- Session 2Dokument47 SeitenSession 2Manish YadavNoch keine Bewertungen
- 4211 Solns 06Dokument7 Seiten4211 Solns 06Roy VeseyNoch keine Bewertungen
- Shock Sensitivity of Explosives Clarified: TechnologyDokument1 SeiteShock Sensitivity of Explosives Clarified: TechnologyNicolás GrinbergNoch keine Bewertungen
- Ioqp 2021 22 Part I SolutionDokument11 SeitenIoqp 2021 22 Part I SolutionJyoti DhillonNoch keine Bewertungen
- Atomic Structure & Interatomic Bonding: Chapter 2 - 1Dokument73 SeitenAtomic Structure & Interatomic Bonding: Chapter 2 - 1dd adminNoch keine Bewertungen
- Lect 02 2020Dokument8 SeitenLect 02 2020Hukry AingNoch keine Bewertungen
- Electron-Electron Interactions: Dragica VasileskaDokument61 SeitenElectron-Electron Interactions: Dragica VasileskaCallum JohnstonNoch keine Bewertungen
- Last Minute Revision Notes ChemistryDokument9 SeitenLast Minute Revision Notes Chemistrytechwithtarun477Noch keine Bewertungen
- University of London: Ph4211A: Statistical MechanicsDokument5 SeitenUniversity of London: Ph4211A: Statistical MechanicsRoy VeseyNoch keine Bewertungen
- Formula Sheet For Astronomy 1 - Paper 1 and Stars & PlanetsDokument2 SeitenFormula Sheet For Astronomy 1 - Paper 1 and Stars & PlanetsprashinNoch keine Bewertungen
- Rubidium87numbers.1.6 Copiar (AutoGuardar)Dokument29 SeitenRubidium87numbers.1.6 Copiar (AutoGuardar)Aldair BernalNoch keine Bewertungen
- D0685 Phy Paper 3Dokument18 SeitenD0685 Phy Paper 3blogeraryanNoch keine Bewertungen
- 2019-SOL-APA-2B - Liquids, POM, Thermodynamics, SHM, WavesDokument12 Seiten2019-SOL-APA-2B - Liquids, POM, Thermodynamics, SHM, WavesHemendra PrasannaNoch keine Bewertungen
- Rubidium 87 NumbersDokument31 SeitenRubidium 87 NumbersLizeth OcampoNoch keine Bewertungen
- Unit 2 Lecture 6 - Energy ConsiderationsDokument9 SeitenUnit 2 Lecture 6 - Energy ConsiderationsEstherNoch keine Bewertungen
- 강의 (2019 03 11) PDFDokument22 Seiten강의 (2019 03 11) PDFHyungryul Daniel YangNoch keine Bewertungen
- Quick Revision Notes ChemistryDokument9 SeitenQuick Revision Notes Chemistrybobby wNoch keine Bewertungen
- CHEL01E 6 - Nuclear Chemistry and EnergyDokument54 SeitenCHEL01E 6 - Nuclear Chemistry and EnergyAnjeline FerrerNoch keine Bewertungen
- Jahnteller Effect Unit 3 2017Dokument15 SeitenJahnteller Effect Unit 3 2017Jaleel BrownNoch keine Bewertungen
- CY1001-2015 Inorganic Lecture NotesDokument16 SeitenCY1001-2015 Inorganic Lecture NotesCreative ThinkerNoch keine Bewertungen
- New Brochure 20pagesDokument20 SeitenNew Brochure 20pagesSikander0% (1)
- Hytrel Product Reference GuideDokument4 SeitenHytrel Product Reference GuideashkansoheylNoch keine Bewertungen
- Combined Gas LawDokument19 SeitenCombined Gas LawDhea Angela A. Capuyan100% (2)
- Sathyabama UniversityDokument20 SeitenSathyabama UniversityMathavaraja JeyaramanNoch keine Bewertungen
- Sephadex LH-20Dokument8 SeitenSephadex LH-20Gustavo Godoy100% (1)
- Types of PropellantDokument3 SeitenTypes of Propellantkartikey0% (1)
- Ibuprofen JP XVIIIDokument1 SeiteIbuprofen JP XVIIIcamilo.carrilloNoch keine Bewertungen
- What Is Lithium CarbonateDokument8 SeitenWhat Is Lithium CarbonateNurAneesaNoch keine Bewertungen
- Chapter 29 Mechanical Separations 18Dokument101 SeitenChapter 29 Mechanical Separations 18Ali HasSsanNoch keine Bewertungen
- Stepan Formulation 1298Dokument2 SeitenStepan Formulation 1298Mohamed AdelNoch keine Bewertungen
- Original Articles / / /: Home Archives Diyala Journal of Engineering Sciences Vol.15, No 3, September 2022Dokument7 SeitenOriginal Articles / / /: Home Archives Diyala Journal of Engineering Sciences Vol.15, No 3, September 2022Celvin SamNoch keine Bewertungen
- Covid Ecri - Disinfectant Concentrations For Epa ListDokument16 SeitenCovid Ecri - Disinfectant Concentrations For Epa ListNikesh ShahNoch keine Bewertungen
- TDS - Pipeclad HOT 120Dokument2 SeitenTDS - Pipeclad HOT 120Long ChenNoch keine Bewertungen
- Design and Analysis of A Gas Turbine Blade by Using FEM: L.UmamaheswararaoDokument6 SeitenDesign and Analysis of A Gas Turbine Blade by Using FEM: L.UmamaheswararaoMohdShahidNoch keine Bewertungen
- All 1314 Chap10-Grand Canonical EnsembleDokument9 SeitenAll 1314 Chap10-Grand Canonical Ensemblegiovanny_francisNoch keine Bewertungen
- Electro NegativityDokument6 SeitenElectro NegativityWarisha AkbarNoch keine Bewertungen
- Bismuth To GoldDokument20 SeitenBismuth To GoldTony Gary67% (3)
- CarcinogensDokument85 SeitenCarcinogensNidyaletchmy ReddyNoch keine Bewertungen
- Assessment Test in Science Module 2Dokument2 SeitenAssessment Test in Science Module 2Caren Mae PunzalanNoch keine Bewertungen
- Prestressed Concrete: BY:-Dr. Mohd Ashraf Iqbal Associate Professor Department of Civil Engineering, IIT, RoorkeeDokument153 SeitenPrestressed Concrete: BY:-Dr. Mohd Ashraf Iqbal Associate Professor Department of Civil Engineering, IIT, RoorkeeAllyson DulfoNoch keine Bewertungen
- 1996 - Kelham - The Effect of Cement Composition and FinenessDokument9 Seiten1996 - Kelham - The Effect of Cement Composition and FinenessRodrigo Henryque Reginato Quevedo Melo100% (1)
- Heater E-501 - MechanicalDokument32 SeitenHeater E-501 - MechanicalLai HuynhNoch keine Bewertungen
- Polarity of MoleculesDokument5 SeitenPolarity of Moleculesgeron pierre BayatanNoch keine Bewertungen
- 2 Texts About Petroleum For Reading ComprehensionDokument4 Seiten2 Texts About Petroleum For Reading ComprehensionEmmanuel Tartagal100% (2)