Das könnte Ihnen auch gefallen
- Medico Legal 1Dokument1 SeiteMedico Legal 1kaye choi75% (8)
- PADI Rescue Diver - Blank Knowledge ReviewDokument13 SeitenPADI Rescue Diver - Blank Knowledge ReviewAj Quek67% (3)
- Local Anaesthetic Doses Guidance - NCH OnlyDokument2 SeitenLocal Anaesthetic Doses Guidance - NCH OnlyakshayNoch keine Bewertungen
- (Shin-Etsu) PHARMACOAT (ENG) - 20140917Dokument16 Seiten(Shin-Etsu) PHARMACOAT (ENG) - 20140917Antonio ReguilloNoch keine Bewertungen
- BioavaibilityDokument20 SeitenBioavaibilityTanChantreaNoch keine Bewertungen
- Poison and Antidote ChartDokument5 SeitenPoison and Antidote ChartSusanne Mae Gonzales50% (2)
- Syndrome of Inappropriate Antidiuretic Hormone Secretion SIADHDokument14 SeitenSyndrome of Inappropriate Antidiuretic Hormone Secretion SIADHbhatubim100% (1)
- Zung Depression Rating Scale PDFDokument2 SeitenZung Depression Rating Scale PDFdienfahrezaNoch keine Bewertungen
- Desi AstiyasariDokument7 SeitenDesi AstiyasariAdelia KhaerunisaNoch keine Bewertungen
- Farm A Codina MicDokument17 SeitenFarm A Codina MicElías ZakzukNoch keine Bewertungen
- ATU V 1.0 February 2020 FinalDokument41 SeitenATU V 1.0 February 2020 FinalMarcelo UGNoch keine Bewertungen
- Pharmaco DynamicsDokument40 SeitenPharmaco DynamicsOssy OsmanNoch keine Bewertungen
- OptiPhos Plus Brochure ENGDokument4 SeitenOptiPhos Plus Brochure ENGdanielleayyash1Noch keine Bewertungen
- PK 1Dokument27 SeitenPK 1Rozzalien BesharaNoch keine Bewertungen
- Second Paper TURKYDokument15 SeitenSecond Paper TURKYmaysam mohammedNoch keine Bewertungen
- Batch Adsorption Process For Phenol Removal From Aqueous Solution Using Walnut Shell ASHDokument20 SeitenBatch Adsorption Process For Phenol Removal From Aqueous Solution Using Walnut Shell ASHکبری ادریس رسولNoch keine Bewertungen
- 09 Nonlinear PKDokument17 Seiten09 Nonlinear PKintanNoch keine Bewertungen
- Open Babylog Gbr61733xxt04Dokument1 SeiteOpen Babylog Gbr61733xxt04MarceloNoch keine Bewertungen
- Pharmacokinetic Drug Interactions: Ika Puspita Sari Bagian Farmakologi & Farmasi Klinik Fakultas Farmasi UGMDokument44 SeitenPharmacokinetic Drug Interactions: Ika Puspita Sari Bagian Farmakologi & Farmasi Klinik Fakultas Farmasi UGMFitrah Craoenk Blauw LiefhebbersNoch keine Bewertungen
- 10 1007@s41742-018Dokument9 Seiten10 1007@s41742-018Radouane El-AmriNoch keine Bewertungen
- Behavior Modification Treatment PhaseDokument5 SeitenBehavior Modification Treatment PhaseMeredith EckardNoch keine Bewertungen
- Dose Response and Concentration Response Analysis 2006-2007Dokument44 SeitenDose Response and Concentration Response Analysis 2006-2007WidnyanaAryawigunaNoch keine Bewertungen
- J. Welty, Et. Al., - Fundamentals of Momentum, Heat and Mass Transfer-Wiley (2008) - 521-526 (1) - 6Dokument1 SeiteJ. Welty, Et. Al., - Fundamentals of Momentum, Heat and Mass Transfer-Wiley (2008) - 521-526 (1) - 6Hugo LacerdaNoch keine Bewertungen
- Bioavailability of DrugsDokument42 SeitenBioavailability of DrugsAnjali TakkeNoch keine Bewertungen
- Bioavailability and BioeqDokument51 SeitenBioavailability and BioeqDhinchakk PoojaNoch keine Bewertungen
- Ulangan 1 Ulangan 1 Ulangan 2: Konsentrasi ( G/ML) Konsentrasi ( G/ML)Dokument1 SeiteUlangan 1 Ulangan 1 Ulangan 2: Konsentrasi ( G/ML) Konsentrasi ( G/ML)sitiNoch keine Bewertungen
- Maximum Recommended Local Anaesthetic Doses For AdultsDokument2 SeitenMaximum Recommended Local Anaesthetic Doses For AdultsadithardanaNoch keine Bewertungen
- Portada: Uanl Nombre Del ProyectoDokument10 SeitenPortada: Uanl Nombre Del ProyectoAydee GarciaNoch keine Bewertungen
- Comparison of Different Techniques For Extraction of Biologically Active Compounds From Achillea Millefolium ProaDokument4 SeitenComparison of Different Techniques For Extraction of Biologically Active Compounds From Achillea Millefolium ProaSandal PutusNoch keine Bewertungen
- PHARMACOKINETICS: What The Body Does To The Drug: - Encompasses The Processes of AbsorptionDokument47 SeitenPHARMACOKINETICS: What The Body Does To The Drug: - Encompasses The Processes of AbsorptionWong Sin Ting HebeNoch keine Bewertungen
- USP-NF Cefotaxime SodiumDokument6 SeitenUSP-NF Cefotaxime SodiumCongluanNoch keine Bewertungen
- CellTiter 96 AQueous One Solution Cell Proliferation Assay TB245Dokument13 SeitenCellTiter 96 AQueous One Solution Cell Proliferation Assay TB245Teh Chye PhingNoch keine Bewertungen
- Problem Set 2Dokument8 SeitenProblem Set 2neekpro2Noch keine Bewertungen
- STK Friday, December 22, 2023Dokument2 SeitenSTK Friday, December 22, 2023Johannus Susanto WibisonoNoch keine Bewertungen
- Doxycycline Prolonged Release CapsulesDokument2 SeitenDoxycycline Prolonged Release CapsulesAlexandra CociuNoch keine Bewertungen
- Chemistry Lab ReportDokument9 SeitenChemistry Lab Reportlwashaba04Noch keine Bewertungen
- 2020 GMI-Webinar COMBINEDDokument61 Seiten2020 GMI-Webinar COMBINEDUntitled TsangNoch keine Bewertungen
- STK 01 April 2023Dokument2 SeitenSTK 01 April 2023Johannus Susanto WibisonoNoch keine Bewertungen
- USP-NF Cefotaxime InjectionDokument3 SeitenUSP-NF Cefotaxime InjectionCongluanNoch keine Bewertungen
- 02 Bioavailability-1Dokument63 Seiten02 Bioavailability-1Toqa ElmansouryNoch keine Bewertungen
- Name That MedicineDokument1 SeiteName That Medicinegolden fleeceNoch keine Bewertungen
- Dose EffectDokument45 SeitenDose Effectapi-19916399Noch keine Bewertungen
- Dose-Effect Relationship Dose-Effect RelationshipDokument43 SeitenDose-Effect Relationship Dose-Effect Relationshipapi-19916399Noch keine Bewertungen
- Yurtlu 2013Dokument14 SeitenYurtlu 2013dana40018256Noch keine Bewertungen
- Pengantar Farmakokinetika: Diana Holidah, Apt., M.Farm. Bag. Farmasi Klinik Dan Komunitas Fakultas Farmasi Univ. JemberDokument33 SeitenPengantar Farmakokinetika: Diana Holidah, Apt., M.Farm. Bag. Farmasi Klinik Dan Komunitas Fakultas Farmasi Univ. JemberfitrinurussaniNoch keine Bewertungen
- 1 s2.0 S0006497120860169 mmc1Dokument6 Seiten1 s2.0 S0006497120860169 mmc1Purba DaripaNoch keine Bewertungen
- Seatwork1 PHARMADokument4 SeitenSeatwork1 PHARMAIzza RojeroNoch keine Bewertungen
- Melilotus ElegansDokument3 SeitenMelilotus ElegansBiniam PaulosNoch keine Bewertungen
- Match Game Matching Statistics & Graphs: What Do I Do?Dokument3 SeitenMatch Game Matching Statistics & Graphs: What Do I Do?Isabella Valencia VernazaNoch keine Bewertungen
- USP-NF Abiraterone Acetate TabletsDokument5 SeitenUSP-NF Abiraterone Acetate Tabletsmustafa bNoch keine Bewertungen
- Thinkswap Pharmaco Practical Report 1Dokument9 SeitenThinkswap Pharmaco Practical Report 1ESTHER WONG TZE YIING -Noch keine Bewertungen
- Kadar Obat MG/L Vs Waktu (Jam) SemilogaritmikDokument6 SeitenKadar Obat MG/L Vs Waktu (Jam) SemilogaritmikIffah ArfianiNoch keine Bewertungen
- Empagliflozin and Linagliptin 2020 Sept FinalDokument65 SeitenEmpagliflozin and Linagliptin 2020 Sept FinalJenny Calapati TorrijosNoch keine Bewertungen
- TAP BlockDokument27 SeitenTAP BlockSneha PanditNoch keine Bewertungen
- Herbal MedicineDokument40 SeitenHerbal MedicinePutri Anom SariNoch keine Bewertungen
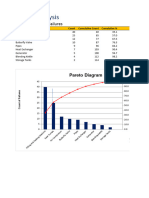
- Pareto Analysis Template 1Dokument2 SeitenPareto Analysis Template 1Georgina SuleNoch keine Bewertungen
- Binks AX260L Air Motor BreakdownDokument11 SeitenBinks AX260L Air Motor BreakdownAryo KamandanuNoch keine Bewertungen
- Eneral Pharmacology: PharmacokineticsDokument36 SeitenEneral Pharmacology: PharmacokineticsSupreet Singh MalhiNoch keine Bewertungen
- Stocktake Maret22Dokument3 SeitenStocktake Maret22RullyNoch keine Bewertungen
- Clinical - Pharmacokinetics 3rd Year BushDokument53 SeitenClinical - Pharmacokinetics 3rd Year BushBaguma MichaelNoch keine Bewertungen
- 11esther Wong Tze Yiing Pharmaco Practical Report 1 PDFDokument9 Seiten11esther Wong Tze Yiing Pharmaco Practical Report 1 PDFESTHER WONG TZE YIING -Noch keine Bewertungen
- Phys 1011labDokument50 SeitenPhys 1011labsemabay71% (7)
- Test Document 4Dokument25 SeitenTest Document 4Ufuk YazganNoch keine Bewertungen
- The Scoop On Brain Healt Dietary Supplement Products Containing Huperzine ADokument7 SeitenThe Scoop On Brain Healt Dietary Supplement Products Containing Huperzine Avasilyi IvanenkoNoch keine Bewertungen
- Death Wish ListDokument6 SeitenDeath Wish ListphilippatstonNoch keine Bewertungen
- (Serbian Journal of Dermatology and Venereology) National Guidelines For The Treatment of Atopic DermatitisDokument25 Seiten(Serbian Journal of Dermatology and Venereology) National Guidelines For The Treatment of Atopic DermatitisFebtri IRnawitaNoch keine Bewertungen
- Kubie, L. (1971) - The Destructive Potential of Humor in PsychotherapyDokument6 SeitenKubie, L. (1971) - The Destructive Potential of Humor in PsychotherapyMikaelaMundell100% (1)
- GBS Review2Dokument64 SeitenGBS Review2Vladimir BasurtoNoch keine Bewertungen
- Lesson Plan in MAPEH (Dagalea)Dokument4 SeitenLesson Plan in MAPEH (Dagalea)Keziah LlenaresNoch keine Bewertungen
- Society For Pediatric Anesthesia Emergency Checklist ManualDokument28 SeitenSociety For Pediatric Anesthesia Emergency Checklist ManualJill SweetNoch keine Bewertungen
- Rectal Prolapse PDFDokument92 SeitenRectal Prolapse PDFadel santos100% (2)
- Death & Dying: and How We Cope With Grief and LossDokument18 SeitenDeath & Dying: and How We Cope With Grief and Losscarlos alemanNoch keine Bewertungen
- List of Medical AbbreviationsDokument16 SeitenList of Medical AbbreviationsClaire Nimor VentulanNoch keine Bewertungen
- Biomedical Acupuncture For Sports and Trauma Rehabilitation: Dry Needling TechniquesDokument6 SeitenBiomedical Acupuncture For Sports and Trauma Rehabilitation: Dry Needling Techniquesbysinuti0% (2)
- Health Checklist Form For Visitors: Nakaranas Ka Ba NG Mga Sumusunod: Oo HindiDokument2 SeitenHealth Checklist Form For Visitors: Nakaranas Ka Ba NG Mga Sumusunod: Oo HindiCoin CharNoch keine Bewertungen
- NCM 113 Community Health Nursing IiDokument4 SeitenNCM 113 Community Health Nursing IiRenee CamilleNoch keine Bewertungen
- Senarai Rakan Strategik KGC As 14.7.2020 - To Put On WebsiteDokument10 SeitenSenarai Rakan Strategik KGC As 14.7.2020 - To Put On WebsiteIvy ChaiNoch keine Bewertungen
- Dengue SIR Model in Baguio (Math197 Paper)Dokument2 SeitenDengue SIR Model in Baguio (Math197 Paper)DreamCatcherNoch keine Bewertungen
- GDMDokument30 SeitenGDMCharlz ZipaganNoch keine Bewertungen
- NiramayaDokument5 SeitenNiramayaIam WaitNoch keine Bewertungen
- SDMS ID: P2010/0496-001 2.9/09WACS Title: Management of Postpartum HaemorrhageDokument11 SeitenSDMS ID: P2010/0496-001 2.9/09WACS Title: Management of Postpartum HaemorrhageYwagar YwagarNoch keine Bewertungen
- Caring For A Nephrostomy Tube at HomeDokument4 SeitenCaring For A Nephrostomy Tube at HomeUmi KrisdyantiniNoch keine Bewertungen
- Tech TriageDokument50 SeitenTech TriagefadiNoch keine Bewertungen
- Nitte News Jan-Dec 2010Dokument88 SeitenNitte News Jan-Dec 2010yvj_20006373Noch keine Bewertungen
- Oral and Maxillofacial Manifestations in Patients With Drug AddictionDokument8 SeitenOral and Maxillofacial Manifestations in Patients With Drug AddictionDiana Mihaela IlieNoch keine Bewertungen
- Prescribing For The ElderlyDokument8 SeitenPrescribing For The ElderlykarladeyNoch keine Bewertungen
- Nurs FPX 4030 Assessment 2 Determining The Credibility of Evidence and ResourcesDokument5 SeitenNurs FPX 4030 Assessment 2 Determining The Credibility of Evidence and ResourcesEmma WatsonNoch keine Bewertungen
- Infection Control Management Plan: PurposeDokument6 SeitenInfection Control Management Plan: PurposeRekhaNoch keine Bewertungen