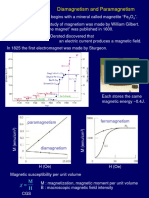
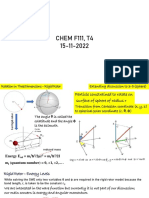
Das könnte Ihnen auch gefallen
- The Intelligent Investor NotesDokument19 SeitenThe Intelligent Investor NotesJack Jacinto100% (6)
- Never Can Say Goodbye Katherine JacksonDokument73 SeitenNever Can Say Goodbye Katherine Jacksonalina28sept100% (5)
- 18 Ex Parte Applic Shorten Time Consolidate 11/01/21Dokument13 Seiten18 Ex Parte Applic Shorten Time Consolidate 11/01/21José DuarteNoch keine Bewertungen
- 1H NMR SpectrosDokument84 Seiten1H NMR Spectrosapi-3723327100% (5)
- Spelling Menu Days and MonthsDokument1 SeiteSpelling Menu Days and MonthsLisl WindhamNoch keine Bewertungen
- Proton Nuclear Magnetic Resonance Spectroscopy H - NMRDokument61 SeitenProton Nuclear Magnetic Resonance Spectroscopy H - NMRchemist100% (2)
- NOTE: Bohr's ModelDokument43 SeitenNOTE: Bohr's ModelmsccenterNoch keine Bewertungen
- A Book of Beasts PDFDokument32 SeitenA Book of Beasts PDFbrad drac100% (2)
- NEET UG Physics Atom and Nucleus MCQsDokument36 SeitenNEET UG Physics Atom and Nucleus MCQsKapila KarthikeyanNoch keine Bewertungen
- Chapter 8 - Nuclear Magnetic Resonance SpectrosDokument38 SeitenChapter 8 - Nuclear Magnetic Resonance SpectrosBizuayehu Gero100% (1)
- Molina Vs de La Riva 6 Phil 12 INOKDokument2 SeitenMolina Vs de La Riva 6 Phil 12 INOKErick Jay InokNoch keine Bewertungen
- Bohr Model of H-AtomDokument18 SeitenBohr Model of H-AtomBhargav100% (1)
- Process Plant Layout - Becoming A Lost ArtDokument7 SeitenProcess Plant Layout - Becoming A Lost ArtRajendraNoch keine Bewertungen
- NMR For BSCDokument13 SeitenNMR For BSCmalluchaithra88_3587Noch keine Bewertungen
- NMR SpectrosDokument66 SeitenNMR SpectrosDushyant Patel100% (1)
- NMRDokument48 SeitenNMRSREEHARI V NAIRNoch keine Bewertungen
- NMR-1-theory and InstrDokument38 SeitenNMR-1-theory and InstrSiddarth PalletiNoch keine Bewertungen
- Introduction 1D 2D NMRDokument23 SeitenIntroduction 1D 2D NMRDelicz TanNoch keine Bewertungen
- NMR 3Dokument29 SeitenNMR 3knkoradiyaNoch keine Bewertungen
- Electron Paramagnetic ResonanceDokument47 SeitenElectron Paramagnetic Resonance150819850Noch keine Bewertungen
- Organic ChemistryDokument101 SeitenOrganic ChemistryHasithaNoch keine Bewertungen
- Belur Zeeman Effect1Dokument20 SeitenBelur Zeeman Effect1nirmalya.sankardasNoch keine Bewertungen
- Chapter 11Dokument43 SeitenChapter 11Neha MohamadNoch keine Bewertungen
- By Syed Ahsan (Upload)Dokument12 SeitenBy Syed Ahsan (Upload)Syed Ahsan Ali ShahNoch keine Bewertungen
- Electronic Configuration NotesDokument1 SeiteElectronic Configuration NotesKraig Andre RellegueNoch keine Bewertungen
- Atomic StructureDokument28 SeitenAtomic StructurePavan GoudNoch keine Bewertungen
- Rotational Raman Spectroscopy: The Polarizability of The Molecule Must Be AnisotropicDokument21 SeitenRotational Raman Spectroscopy: The Polarizability of The Molecule Must Be AnisotropicKartik RanaNoch keine Bewertungen
- Presymposium Workshop Part2 May28Dokument71 SeitenPresymposium Workshop Part2 May28Smruti Snigdha MishraNoch keine Bewertungen
- NMR - 01Dokument49 SeitenNMR - 01Business MatterNoch keine Bewertungen
- Presented by Devisha Tatineni Pharmaceutical Chemistry M.PharmDokument222 SeitenPresented by Devisha Tatineni Pharmaceutical Chemistry M.PharmRashed BiswasNoch keine Bewertungen
- Excited Nucleon Electromagnetic Form Factors From Broken Spin-Flavor SymmetryDokument52 SeitenExcited Nucleon Electromagnetic Form Factors From Broken Spin-Flavor SymmetrySuman DuttaNoch keine Bewertungen
- Kuliah NMRDokument92 SeitenKuliah NMRDedi saputraNoch keine Bewertungen
- 02 08II. Electromagnetic Induction 215-225Dokument7 Seiten02 08II. Electromagnetic Induction 215-225eamcetmaterials0% (1)
- Applications of H, C, F and P - NMR Spectroscopy in The Structural Assessment of Inorganic CompoundsDokument25 SeitenApplications of H, C, F and P - NMR Spectroscopy in The Structural Assessment of Inorganic CompoundsSaurav PaulNoch keine Bewertungen
- Zeeman Effect: 1 Aim of The ExperimentDokument6 SeitenZeeman Effect: 1 Aim of The ExperimentAmlandeep NayakNoch keine Bewertungen
- ZeemanDokument4 SeitenZeemanBhabeshSarangiNoch keine Bewertungen
- ESR MSC PreviousDokument23 SeitenESR MSC PreviousMeenakshiputraeashwarprasad MacherlaNoch keine Bewertungen
- Chapter Eleven Diamagnetism andDokument43 SeitenChapter Eleven Diamagnetism and許良兆Noch keine Bewertungen
- Electron Spin ResonanceDokument8 SeitenElectron Spin ResonanceGaluh PrameswariNoch keine Bewertungen
- R Sum AtomedeBohr-enDokument5 SeitenR Sum AtomedeBohr-enmilina moliNoch keine Bewertungen
- JEE Main 2021 17 March Shift 1 PhysicsDokument13 SeitenJEE Main 2021 17 March Shift 1 PhysicsAditya Raj SinghNoch keine Bewertungen
- Lecture 03Dokument12 SeitenLecture 03Mohit RawatNoch keine Bewertungen
- Aakash JeeDokument5 SeitenAakash JeeVansh BhardwajNoch keine Bewertungen
- Bohr's Model of The AtomDokument8 SeitenBohr's Model of The AtomMuhammad Awais TariqNoch keine Bewertungen
- Interference of Light Waves: ObjectivesDokument25 SeitenInterference of Light Waves: Objectivesjay singhviNoch keine Bewertungen
- Antiaromatic Dianion (Aromatic) : (12) - AnnuleneDokument22 SeitenAntiaromatic Dianion (Aromatic) : (12) - AnnuleneRashmi AgrawalNoch keine Bewertungen
- B.SC - Ii Paper-B (Optics and Lasers) : Submitted by Dr. Sarvpreet Kaur Assistant Professor PGGCG-11, ChandigarhDokument26 SeitenB.SC - Ii Paper-B (Optics and Lasers) : Submitted by Dr. Sarvpreet Kaur Assistant Professor PGGCG-11, ChandigarhAanchal SarwanNoch keine Bewertungen
- Nuclear Magnetic Resonance (NMR) SpectrosDokument47 SeitenNuclear Magnetic Resonance (NMR) SpectrosFrancisco Javier Escobar MedinaNoch keine Bewertungen
- Spectroscopy (L-26 To 29)Dokument81 SeitenSpectroscopy (L-26 To 29)Vashistha GargNoch keine Bewertungen
- Ch4 Fine Structure of AtomsDokument17 SeitenCh4 Fine Structure of AtomsAyorinde T TundeNoch keine Bewertungen
- BITS Pilani Chemistry Hamilton OperatorDokument11 SeitenBITS Pilani Chemistry Hamilton OperatorNaresh SehdevNoch keine Bewertungen
- Esr 1Dokument18 SeitenEsr 1TamizhNoch keine Bewertungen
- CHM 101 - 2Dokument8 SeitenCHM 101 - 2enocholadeinde2004Noch keine Bewertungen
- Modern Physics For Scientists and Engineers 4Th Edition Thornton Solutions Manual Full Chapter PDFDokument31 SeitenModern Physics For Scientists and Engineers 4Th Edition Thornton Solutions Manual Full Chapter PDFteniasisrehearsex8ei100% (9)
- Final Neet (Ug) - 2023 Phase 2 (Manipur Examination) Test Paper With Answer & Detailed Solution (Held On Tuesday 6th JUNE, 2023)Dokument37 SeitenFinal Neet (Ug) - 2023 Phase 2 (Manipur Examination) Test Paper With Answer & Detailed Solution (Held On Tuesday 6th JUNE, 2023)aiimstarget2024Noch keine Bewertungen
- NMRDokument173 SeitenNMRঋ ত্বিকNoch keine Bewertungen
- Chemistry 1A Spectroscopy.: Prof. Mike Ashfold (S305) (Mike - Ashfold@bris - Ac.uk)Dokument63 SeitenChemistry 1A Spectroscopy.: Prof. Mike Ashfold (S305) (Mike - Ashfold@bris - Ac.uk)Abd El-Fattah Mohamed OufNoch keine Bewertungen
- Atomic and Nuclear Physics: Electron Spin Resonance at DPPHDokument6 SeitenAtomic and Nuclear Physics: Electron Spin Resonance at DPPHAlejandra AwimbaweNoch keine Bewertungen
- NMR Spectroscopy: by Darshan R. Telange, KNCP, Butibori (Nagpur)Dokument60 SeitenNMR Spectroscopy: by Darshan R. Telange, KNCP, Butibori (Nagpur)team engineerNoch keine Bewertungen
- BemstrahlungDokument15 SeitenBemstrahlungPrizm Rose'sNoch keine Bewertungen
- Notes 05 HMR v26 Part1Dokument38 SeitenNotes 05 HMR v26 Part1cj AndersonNoch keine Bewertungen
- NMR 1 PDFDokument53 SeitenNMR 1 PDFartaNoch keine Bewertungen
- Collective ModelDokument22 SeitenCollective ModelYogendra MeenaNoch keine Bewertungen
- Nuclear Magnetic Resonance (NMR) SpectrosDokument52 SeitenNuclear Magnetic Resonance (NMR) Spectrossharifah sakinah syed soffianNoch keine Bewertungen
- Atomic Short NotesDokument2 SeitenAtomic Short NotesUiiNoch keine Bewertungen
- Tables of Coefficients for the Analysis of Triple Angular Correlations of Gamma-Rays from Aligned NucleiVon EverandTables of Coefficients for the Analysis of Triple Angular Correlations of Gamma-Rays from Aligned NucleiNoch keine Bewertungen
- The Stopping and Ranges of Ions in Matter: Handbook of Stopping Cross-Sections for Energetic Ions in All ElementsVon EverandThe Stopping and Ranges of Ions in Matter: Handbook of Stopping Cross-Sections for Energetic Ions in All ElementsNoch keine Bewertungen
- 1.Tandem in space (空間性串聯) 2.Tandem in time (時間性串聯)Dokument14 Seiten1.Tandem in space (空間性串聯) 2.Tandem in time (時間性串聯)api-3696530Noch keine Bewertungen
- 10 09 2006 EcologyDokument16 Seiten10 09 2006 Ecologyapi-3696530Noch keine Bewertungen
- 電生理Dokument2 Seiten電生理api-3696530Noch keine Bewertungen
- Ecology 2 NDUDokument19 SeitenEcology 2 NDUapi-3696530Noch keine Bewertungen
- Antibody Microarrays: Microarray FabricationDokument15 SeitenAntibody Microarrays: Microarray Fabricationapi-3696530Noch keine Bewertungen
- EcologyDokument15 SeitenEcologyapi-3696530Noch keine Bewertungen
- CMSC 838T - Lecture 9: Bioinformatics DatabasesDokument65 SeitenCMSC 838T - Lecture 9: Bioinformatics Databasesapi-3696530Noch keine Bewertungen
- 13Dokument15 Seiten13api-3696530Noch keine Bewertungen
- LC-LC-MS-MS For Complex Mixtures: MS/MS Method Using ESIDokument20 SeitenLC-LC-MS-MS For Complex Mixtures: MS/MS Method Using ESIapi-3696530Noch keine Bewertungen
- 11Dokument74 Seiten11api-3696530Noch keine Bewertungen
- 2D Gel DatabasesDokument17 Seiten2D Gel Databasesapi-3696530Noch keine Bewertungen
- 12 22 2006 MCB Cell To Cell Signaling 2 (DRDokument48 Seiten12 22 2006 MCB Cell To Cell Signaling 2 (DRapi-3696530Noch keine Bewertungen
- 14Dokument29 Seiten14api-3696530Noch keine Bewertungen
- Library Construction: Cut Into PiecesDokument13 SeitenLibrary Construction: Cut Into Piecesapi-3696530Noch keine Bewertungen
- Vacuum System in MALDI-TOF MSDokument4 SeitenVacuum System in MALDI-TOF MSapi-3696530Noch keine Bewertungen
- 08Dokument17 Seiten08api-3696530Noch keine Bewertungen
- 09Dokument14 Seiten09api-3696530Noch keine Bewertungen
- De Novo Sequencing by Tandem Mass Spectrometry (MS-MS)Dokument10 SeitenDe Novo Sequencing by Tandem Mass Spectrometry (MS-MS)api-3696530Noch keine Bewertungen
- Protein Profiling of Serum in AIDS PatientsDokument6 SeitenProtein Profiling of Serum in AIDS Patientsapi-3696530Noch keine Bewertungen
- 06Dokument48 Seiten06api-3696530Noch keine Bewertungen
- Mass Spectrometry: J. J. Thomson Carriers of Negative Electricity Nobel Lecture, December 11, 1906Dokument14 SeitenMass Spectrometry: J. J. Thomson Carriers of Negative Electricity Nobel Lecture, December 11, 1906api-3696530Noch keine Bewertungen
- 01 08 2007 MCB Cell DifferentiationDokument60 Seiten01 08 2007 MCB Cell Differentiationapi-3696530Noch keine Bewertungen
- 12 29 2006 MCB ch15Dokument85 Seiten12 29 2006 MCB ch15api-3696530Noch keine Bewertungen
- 2D Electrophoresis: LysisDokument15 Seiten2D Electrophoresis: Lysisapi-3696530Noch keine Bewertungen
- DNA Protein: DNA Sequencing Protein Sequencing DNA Fingerprint (Restriction Map) Protein FingerprintDokument13 SeitenDNA Protein: DNA Sequencing Protein Sequencing DNA Fingerprint (Restriction Map) Protein Fingerprintapi-3696530Noch keine Bewertungen
- 12-25-2006-MCB-Hsueh YPDokument16 Seiten12-25-2006-MCB-Hsueh YPapi-3696530Noch keine Bewertungen
- Molecular Cell Biology: Cell To Cell Signaling (I)Dokument41 SeitenMolecular Cell Biology: Cell To Cell Signaling (I)api-3696530Noch keine Bewertungen
- 12 08 2006 MCB Cytoskeleton and Movement II 1Dokument129 Seiten12 08 2006 MCB Cytoskeleton and Movement II 1api-3696530Noch keine Bewertungen
- 12-25-2006-MCB-Hsueh YPDokument16 Seiten12-25-2006-MCB-Hsueh YPapi-3696530Noch keine Bewertungen
- 12-15-2006-MCB-Hsueh YPDokument14 Seiten12-15-2006-MCB-Hsueh YPapi-3696530100% (2)
- Energizing Your ScalesDokument3 SeitenEnergizing Your ScalesjohnNoch keine Bewertungen
- Seerat Mujaddid Alf-e-Sani (Urdu)Dokument518 SeitenSeerat Mujaddid Alf-e-Sani (Urdu)Talib Ghaffari100% (12)
- TestFunda - Puzzles 1Dokument39 SeitenTestFunda - Puzzles 1Gerald KohNoch keine Bewertungen
- Sec 25 HmaDokument3 SeitenSec 25 HmaMukul BajajNoch keine Bewertungen
- Men Orr HagiaDokument8 SeitenMen Orr HagiaAtika Bashirati IlmanNoch keine Bewertungen
- Management of Breast CancerDokument53 SeitenManagement of Breast CancerGaoudam NatarajanNoch keine Bewertungen
- Luzande, Mary Christine B - Motivating and Managing Individuals - Moral LeadershipDokument15 SeitenLuzande, Mary Christine B - Motivating and Managing Individuals - Moral LeadershipMAry Christine BatongbakalNoch keine Bewertungen
- Physical Education 10 WEEK 2Dokument10 SeitenPhysical Education 10 WEEK 2Israel MarquezNoch keine Bewertungen
- Speakout Language BankDokument7 SeitenSpeakout Language BankСаша БулуєвNoch keine Bewertungen
- Final DemoDokument14 SeitenFinal DemoangieNoch keine Bewertungen
- Persuasive SpeechDokument2 SeitenPersuasive SpeechAngel Zachary RayyanNoch keine Bewertungen
- Brand Zara GAP Forever 21 Mango H&M: Brand Study of Zara Nancys Sharma FD Bdes Batch 2 Sem 8 Brand-ZaraDokument2 SeitenBrand Zara GAP Forever 21 Mango H&M: Brand Study of Zara Nancys Sharma FD Bdes Batch 2 Sem 8 Brand-ZaraNancy SharmaNoch keine Bewertungen
- CabillanDokument12 SeitenCabillanvivivioletteNoch keine Bewertungen
- Iver Brevik, Olesya Gorbunova and Diego Saez-Gomez - Casimir Effects Near The Big Rip Singularity in Viscous CosmologyDokument7 SeitenIver Brevik, Olesya Gorbunova and Diego Saez-Gomez - Casimir Effects Near The Big Rip Singularity in Viscous CosmologyDex30KMNoch keine Bewertungen
- E-Gift Shopper - Proposal - TemplateDokument67 SeitenE-Gift Shopper - Proposal - TemplatetatsuNoch keine Bewertungen
- Accenture 172199U SAP S4HANA Conversion Brochure US Web PDFDokument8 SeitenAccenture 172199U SAP S4HANA Conversion Brochure US Web PDFrajesh2kakkasseryNoch keine Bewertungen
- Cost Accounting - Course Study Guide. (Repaired)Dokument9 SeitenCost Accounting - Course Study Guide. (Repaired)syed Hassan100% (1)
- SunEdison Pancho Perez Complaint As FiledDokument47 SeitenSunEdison Pancho Perez Complaint As FiledLizHoffmanNoch keine Bewertungen
- Blood Is A Body Fluid in Human and Other Animals That Delivers Necessary Substances Such AsDokument24 SeitenBlood Is A Body Fluid in Human and Other Animals That Delivers Necessary Substances Such AsPaulo DanielNoch keine Bewertungen
- How To Manage Asthma: A GuideDokument44 SeitenHow To Manage Asthma: A GuideSrinivas YerriboinaNoch keine Bewertungen
- 17PME328E: Process Planning and Cost EstimationDokument48 Seiten17PME328E: Process Planning and Cost EstimationDeepak MisraNoch keine Bewertungen
- 21st Century NotesDokument3 Seiten21st Century NotesCarmen De HittaNoch keine Bewertungen
- USA V Rowland - Opposition To Motion To End Probation EarlyDokument12 SeitenUSA V Rowland - Opposition To Motion To End Probation EarlyFOX 61 WebstaffNoch keine Bewertungen