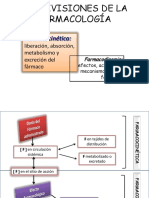
Das könnte Ihnen auch gefallen
- Farmacocinética DrSalasDokument98 SeitenFarmacocinética DrSalasCesar Del Aguila MattaNoch keine Bewertungen
- Farmacocinética y metabolismo de fármacosDokument48 SeitenFarmacocinética y metabolismo de fármacosTaller De Tesis Farmacia UladechNoch keine Bewertungen
- Farmacocinética FP ActualizadoDokument47 SeitenFarmacocinética FP ActualizadoheleanyNoch keine Bewertungen
- Introducción a la farmacocinética clínicaDokument36 SeitenIntroducción a la farmacocinética clínicaTaller De Tesis Farmacia UladechNoch keine Bewertungen
- Teoricos y Talleres 2023Dokument1.117 SeitenTeoricos y Talleres 2023Aldana Jazmin FrancoNoch keine Bewertungen
- 1 Procesos LADME 1Dokument37 Seiten1 Procesos LADME 1Rachelle CorralNoch keine Bewertungen
- FARMACOLOGIA Y TERAPEUTICA VETERINARIA I.ppt 11Dokument197 SeitenFARMACOLOGIA Y TERAPEUTICA VETERINARIA I.ppt 11Adolfo Canaza T100% (1)
- Práctica N°4 - Factores FarmacocinéticosDokument6 SeitenPráctica N°4 - Factores FarmacocinéticosRUTH GABRIELA BENAVIDEZ CARBAJALNoch keine Bewertungen
- FarmacocinéticaDokument54 SeitenFarmacocinéticaJean Pierre Cano MolinaNoch keine Bewertungen
- FARMACOCINETICADokument23 SeitenFARMACOCINETICAPARI MAYTA SARAH EVELYNNoch keine Bewertungen
- FarmacocinticaDokument23 SeitenFarmacocinticaWILDER NUÑUVERO ARCOSNoch keine Bewertungen
- FarmacocineticaDokument23 SeitenFarmacocineticaMargot Mendoza SalasNoch keine Bewertungen
- 2 - Farmacocinetica 2011-IDokument59 Seiten2 - Farmacocinetica 2011-IKatherine LazoNoch keine Bewertungen
- PsicofarmacologíaDokument66 SeitenPsicofarmacologíafiel2003Noch keine Bewertungen
- Farmacocinetica y FarmacodinamiaDokument37 SeitenFarmacocinetica y FarmacodinamiaZulma Madeline L. Gutarra TicaNoch keine Bewertungen
- Teoria 3Dokument33 SeitenTeoria 3HECTOR HEKERIN LUCAS TELLONoch keine Bewertungen
- Farmacología Farmacocinetica Introd UJAPDokument27 SeitenFarmacología Farmacocinetica Introd UJAPAndrea GuiaNoch keine Bewertungen
- Farmacocinética y FarmacodinamiaDokument54 SeitenFarmacocinética y FarmacodinamiaJocelyn AlejandraNoch keine Bewertungen
- Conceptos clave de farmacocinética enDokument23 SeitenConceptos clave de farmacocinética enMarioCraft 15Noch keine Bewertungen
- Mecanismos de Absorcion y Transporte de FarmacosDokument8 SeitenMecanismos de Absorcion y Transporte de FarmacosMiguel AngelNoch keine Bewertungen
- FARMACOCINETICA y FARMACODINAMIADokument25 SeitenFARMACOCINETICA y FARMACODINAMIANadia MoscaNoch keine Bewertungen
- Farmacocinetica y FarmacodinamiaDokument41 SeitenFarmacocinetica y Farmacodinamiabetoasd95Noch keine Bewertungen
- Farmacia (Conceptos Básicos)Dokument46 SeitenFarmacia (Conceptos Básicos)DarkVader9183% (6)
- Farmacodinamia y FarmacocineticaDokument37 SeitenFarmacodinamia y FarmacocineticaJohan JuarezNoch keine Bewertungen
- FarmacocineticaDokument61 SeitenFarmacocineticaalvaro alejandro restrepo zapataNoch keine Bewertungen
- Farmacocintica 2Dokument22 SeitenFarmacocintica 2yuriNoch keine Bewertungen
- Farm Acolo GiaDokument151 SeitenFarm Acolo GiaAldrin López JesúsNoch keine Bewertungen
- Clase Farmacologia General WebDokument92 SeitenClase Farmacologia General WebAgrotienda VasquezNoch keine Bewertungen
- Calculo y Dilucion de MedicamentosDokument39 SeitenCalculo y Dilucion de MedicamentosKarla HernándezNoch keine Bewertungen
- FarmacologíaDokument24 SeitenFarmacologíaNicole OstrovskyNoch keine Bewertungen
- Farmacologia General (Azael)Dokument76 SeitenFarmacologia General (Azael)maryNoch keine Bewertungen
- Resumen de Farmacología GeneralDokument7 SeitenResumen de Farmacología GeneralPatricioRiosCayo100% (1)
- Farmacolog-A B-SicaDokument14 SeitenFarmacolog-A B-SicaDANIELNoch keine Bewertungen
- Farmacocinética y farmacodinamia: procesos de las drogas en el organismoDokument94 SeitenFarmacocinética y farmacodinamia: procesos de las drogas en el organismoReyes BerthaNoch keine Bewertungen
- Farmacocinetica y Farmacodinamia 2Dokument40 SeitenFarmacocinetica y Farmacodinamia 2Micaela Munayco SilvaNoch keine Bewertungen
- Fenómeno Primer PasoDokument3 SeitenFenómeno Primer PasoSory Buele73% (11)
- Absorción, Distribuciòn, Metabolismo y Excreción deDokument53 SeitenAbsorción, Distribuciòn, Metabolismo y Excreción deCarmen LaresNoch keine Bewertungen
- Apuntes de FarmacologíaDokument23 SeitenApuntes de FarmacologíaVan QuiqueNoch keine Bewertungen
- Diapo Clase 2Dokument23 SeitenDiapo Clase 2Anna KareninaNoch keine Bewertungen
- 4.farmacocinetica y Farmacodinamica PDFDokument66 Seiten4.farmacocinetica y Farmacodinamica PDFEvelyn Del Pilar Orellano Pastor100% (1)
- Clase 01 - Introduccion A La FarmacocineticaDokument34 SeitenClase 01 - Introduccion A La FarmacocineticaFlor Lozano JulianNoch keine Bewertungen
- LADMEDokument23 SeitenLADMEdeymellNoch keine Bewertungen
- Fármaco receptorDokument9 SeitenFármaco receptorGeraldine Hernandez FranciscoNoch keine Bewertungen
- Farmacocinética y farmacodinamiaDokument8 SeitenFarmacocinética y farmacodinamiaDiana García JaraNoch keine Bewertungen
- FARMACOLOGIA TP NRO1Dokument8 SeitenFARMACOLOGIA TP NRO1Alexis AvilaNoch keine Bewertungen
- FarmacocinéticaDokument62 SeitenFarmacocinéticaEddy Romario Vilcanqui JiménezNoch keine Bewertungen
- Tema 02. PK - PD - EduardoDokument46 SeitenTema 02. PK - PD - EduardoRAFAEL LUCDÓVICO LIZARDO LEYVANoch keine Bewertungen
- Adme y Parámetros PK 2022Dokument56 SeitenAdme y Parámetros PK 2022Sl SantyNoch keine Bewertungen
- FARMACOLOGÍA Primera Semana PDFDokument180 SeitenFARMACOLOGÍA Primera Semana PDFYamilka Enciso ContrerasNoch keine Bewertungen
- CLASE 3 Farmacocinética I-1Dokument30 SeitenCLASE 3 Farmacocinética I-1LEO100% (1)
- Farmacocinética: procesos de absorción, distribución, metabolismo y eliminaciónDokument59 SeitenFarmacocinética: procesos de absorción, distribución, metabolismo y eliminaciónAlesandra Azurin IcazaNoch keine Bewertungen
- Clase 4 Definicióndelos Diferentes Términos Aplicados en FarmacoterapiaDokument16 SeitenClase 4 Definicióndelos Diferentes Términos Aplicados en FarmacoterapiaKarina MoralesNoch keine Bewertungen
- Diap Proceso Ladme ViasDokument37 SeitenDiap Proceso Ladme ViasPaulo André Maldonado LomelíNoch keine Bewertungen
- Farmacología general: apuntes sobre la ley 20.724, ficha farmacológica y características de los fármacosDokument19 SeitenFarmacología general: apuntes sobre la ley 20.724, ficha farmacológica y características de los fármacosjuan carlosNoch keine Bewertungen
- Farmacocinética I: Absorción, Distribución, Metabolismo y Excreción de FármacosDokument44 SeitenFarmacocinética I: Absorción, Distribución, Metabolismo y Excreción de FármacosAlexaB.MendozaNoch keine Bewertungen
- Pregubtas Del CAPITULO5Dokument3 SeitenPregubtas Del CAPITULO5Tracey Yahsiel Fernandez HidalgoNoch keine Bewertungen
- Farmacocin TicaDokument22 SeitenFarmacocin TicaJulio Cesar Alonso CastroNoch keine Bewertungen
- Farmacologia Semana 2 UnwDokument34 SeitenFarmacologia Semana 2 UnwJade Nicel Liliana Esquivel MogollonNoch keine Bewertungen
- Bases Anatomopatológicas De La Enfermedad Quirúrgica: Tomo IVon EverandBases Anatomopatológicas De La Enfermedad Quirúrgica: Tomo INoch keine Bewertungen
- Nuevos usos para viejos medicamentosVon EverandNuevos usos para viejos medicamentosNoch keine Bewertungen
- MetabolismoDokument33 SeitenMetabolismoapi-371018650% (2)
- Clase - Enzimas - Bioquímica IDokument46 SeitenClase - Enzimas - Bioquímica Iapi-3710186Noch keine Bewertungen
- Clase Región Pelvis AnatomiaDokument29 SeitenClase Región Pelvis Anatomiaapi-3707147100% (2)
- FosforilacionDokument63 SeitenFosforilacionapi-3710186Noch keine Bewertungen
- Clase005 - Carbohidratos - Bioquímica IDokument57 SeitenClase005 - Carbohidratos - Bioquímica Iapi-3710186Noch keine Bewertungen
- Clase - Agua - Bioquímica IDokument25 SeitenClase - Agua - Bioquímica Iapi-3710186Noch keine Bewertungen
- Clase006 - Glicolisis - Bioquímica IDokument43 SeitenClase006 - Glicolisis - Bioquímica Iapi-3710186Noch keine Bewertungen
- Clase - Proteinas2 - Bioquímica IDokument18 SeitenClase - Proteinas2 - Bioquímica Iapi-3710186Noch keine Bewertungen
- Clase007 - Pentosas y Ciclo de Krebs - Bioquímica IDokument54 SeitenClase007 - Pentosas y Ciclo de Krebs - Bioquímica Iapi-3710186Noch keine Bewertungen
- 001 FarmacodinámiaDokument1 Seite001 Farmacodinámiaapi-3710186Noch keine Bewertungen
- Clase - Ingeniería de ProteinasDokument39 SeitenClase - Ingeniería de Proteinasapi-3710186100% (1)
- Clase - Proteinas1 - Bioquímica IDokument30 SeitenClase - Proteinas1 - Bioquímica Iapi-3710186Noch keine Bewertungen
- Exposición - Priones - Introducción A Bioquímica IIDokument14 SeitenExposición - Priones - Introducción A Bioquímica IIapi-3710186100% (1)
- 002 FarmacodinámiaDokument1 Seite002 Farmacodinámiaapi-3710186Noch keine Bewertungen
- Clase003 Receptores Farmacológicos FarmacodinámiaDokument42 SeitenClase003 Receptores Farmacológicos Farmacodinámiaapi-3710186100% (2)
- Exposición - Proyecto Genoma Humano - Introducción A Bioquímica IIDokument12 SeitenExposición - Proyecto Genoma Humano - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Clase006 Sistema Autonómico FarmacodinámiaDokument67 SeitenClase006 Sistema Autonómico Farmacodinámiaapi-3710186100% (1)
- Exposición - Síndrome Vangierke - Introducción A Bioquímica IIDokument17 SeitenExposición - Síndrome Vangierke - Introducción A Bioquímica IIapi-3710186100% (2)
- Clase002 Vida Media FarmacodinámiaDokument47 SeitenClase002 Vida Media Farmacodinámiaapi-3710186100% (1)
- Exposición - VIH - Introducción A Bioquímica IIDokument11 SeitenExposición - VIH - Introducción A Bioquímica IIapi-3710186100% (1)
- Exposición - Porfíria - Introducción A Bioquímica IIDokument12 SeitenExposición - Porfíria - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Exposición - Pelagra - Introducción A Bioquímica IIDokument21 SeitenExposición - Pelagra - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Exposición - Neurotrasmisores - Introducción A Bioquímica IIDokument23 SeitenExposición - Neurotrasmisores - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Exposición - Óxido Nítrico - Introducción A Bioquímica IIDokument15 SeitenExposición - Óxido Nítrico - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Exposición - Parkinson - Introducción A Bioquímica IIDokument18 SeitenExposición - Parkinson - Introducción A Bioquímica IIapi-3710186100% (3)
- Exposición - Intolerancia A La Lactosa - Introducción A Bioquímica IIDokument11 SeitenExposición - Intolerancia A La Lactosa - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Exposición - Fibrósis Quística - Introducción A Bioquímica IIDokument10 SeitenExposición - Fibrósis Quística - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Exposición - Leptina - Introducción A Bioquímica IIDokument14 SeitenExposición - Leptina - Introducción A Bioquímica IIapi-3710186100% (1)
- Exposición - Colesterol - Introducción A Bioquímica IIDokument11 SeitenExposición - Colesterol - Introducción A Bioquímica IIapi-3710186Noch keine Bewertungen
- Tecnología Del Concreto 11.06.15Dokument147 SeitenTecnología Del Concreto 11.06.15Cristian CcNoch keine Bewertungen
- Paper 1 Conductimetría (1) .En - EsDokument9 SeitenPaper 1 Conductimetría (1) .En - EsJohn ArceNoch keine Bewertungen
- Acido TanicoDokument13 SeitenAcido TanicoSh4Dow FoXNoch keine Bewertungen
- Catalogo GD ToolsDokument148 SeitenCatalogo GD ToolstornomanNoch keine Bewertungen
- Poli FarmaciaDokument22 SeitenPoli FarmaciaNely RodríguezNoch keine Bewertungen
- Informe LombriculturaDokument17 SeitenInforme LombriculturaYeison Javier Rodriguez VelaNoch keine Bewertungen
- Memoria de Plan de Negocio Proyecto, Pinos Maderables en La Zona de Queretaro.Dokument48 SeitenMemoria de Plan de Negocio Proyecto, Pinos Maderables en La Zona de Queretaro.Wilnaí Ra-HaraktysNoch keine Bewertungen
- AFFF 50LtsDokument1 SeiteAFFF 50Ltsjose zapataNoch keine Bewertungen
- Informe Mantenimeinto de Obras CivilesDokument7 SeitenInforme Mantenimeinto de Obras CivilesAylín Colina JordánNoch keine Bewertungen
- Resistencia A La Compresión Por CuradoDokument13 SeitenResistencia A La Compresión Por CuradoWalter Jose Lizarraga AvilaNoch keine Bewertungen
- Examen Feb 2015 Upt BiofisicaDokument9 SeitenExamen Feb 2015 Upt BiofisicaLuis Alberto Mendoza SalasNoch keine Bewertungen
- Calentadores Clasic 13 LDokument67 SeitenCalentadores Clasic 13 Ltitan_krakenNoch keine Bewertungen
- Reconocimiento Materiales Acrilicos Resinas AutocuradoDokument7 SeitenReconocimiento Materiales Acrilicos Resinas AutocuradoZoilo Puma QuicañoNoch keine Bewertungen
- Anuario IPA 2006Dokument273 SeitenAnuario IPA 2006Pau Zalazar100% (1)
- La Importancia Del Estudio de La Estequiometría y Su Aplicablilidad en La IndustriaDokument4 SeitenLa Importancia Del Estudio de La Estequiometría y Su Aplicablilidad en La IndustriaBui Quang Hieu100% (1)
- Tabla periódica de los elementosDokument10 SeitenTabla periódica de los elementosElsy Maria Moreno BraqueNoch keine Bewertungen
- Analisis Instrumental, SeguimientoDokument4 SeitenAnalisis Instrumental, SeguimientoDAYRA NATHALY HUERTAS RAMÍREZNoch keine Bewertungen
- Fórmula para Fabricar ShampooDokument3 SeitenFórmula para Fabricar ShampooAdrian CamacaroNoch keine Bewertungen
- BIO021 + AcetilcolinaDokument129 SeitenBIO021 + AcetilcolinaROCIO SINAI HERNANDEZ JUAREZNoch keine Bewertungen
- Toma Lateral - CalculoDokument8 SeitenToma Lateral - Calculocuy18Noch keine Bewertungen
- Ensayos destructivos con varios porcentajes de carbonoDokument10 SeitenEnsayos destructivos con varios porcentajes de carbonojulioNoch keine Bewertungen
- Sistema Hcci de MazdaDokument3 SeitenSistema Hcci de Mazdaleonardodaniel1994Noch keine Bewertungen
- Evaluación Biología 2do BGUDokument3 SeitenEvaluación Biología 2do BGUfranciscoNoch keine Bewertungen
- Rehidratación oral guíaDokument23 SeitenRehidratación oral guíaJimena Rodríguez Soto50% (2)
- Vertederos Rectangulares de Paredes DelgadasDokument2 SeitenVertederos Rectangulares de Paredes DelgadasDiego SalazarNoch keine Bewertungen
- L. Ley de Bouguer Lambert Beer 0 PDFDokument2 SeitenL. Ley de Bouguer Lambert Beer 0 PDFYeisyPamelaZapataCordovaNoch keine Bewertungen
- Reducción cetona alcohol mediante cincDokument3 SeitenReducción cetona alcohol mediante cincCarlos CastilloNoch keine Bewertungen
- Física y Química 2º Eso Primer TrimestreDokument2 SeitenFísica y Química 2º Eso Primer TrimestreenkarniNoch keine Bewertungen
- Seminario Vancomicina 1.3Dokument48 SeitenSeminario Vancomicina 1.3Herbert Martin Colan TorresNoch keine Bewertungen
- Primer Webinar, Frutos Rojos.Dokument3 SeitenPrimer Webinar, Frutos Rojos.Vania GonzalezNoch keine Bewertungen